

Czy cholesterol w układzie krążenia powoduje choroby układu krążenia?
A w zasadzie: czy kardiologia ma rację?

*Tekst nie stanowi dla nikogo żadnej porady medycznej i nie może być podstawą dla kogokolwiek do przerywania lub podejmowania jakiejkolwiek farmakoterapii, która powinna być konsultowana ze specjalistami.
Tekst jest również dostępny w formie prezentacji na Youtube.
Spis treści:
3. Próba falsyfikacji
1. Wprowadzenie w temat cholesterolu i lipidogramu
Wolałbym od razu przejść do odpowiedzi na najbardziej nas nurtujące pytanie, ale niestety nie jest to możliwe z powodu pewnego zamieszania terminologicznego wokół „cholesterolu”. Co więcej, istnieje nawet pewien rozziew między laboratoryjnymi testami, praktyką medyczną a publikacjami naukowymi w tym zakresie. Dlatego nie mam za bardzo wyjścia i muszę zacząć od pewnego mozolnego, momentami nudnego wprowadzenia. Jeśli ktoś nie jest nim bezpośrednio zainteresowany, lub zna klasyfikacje, to może przejść od razu do sekcji drugiej o interesującej nas hipotezie, albo sekcji trzeciej o jej próbie falsyfikacji.
Cholesterol w organizmie, diecie i krążeniu
To opracowanie będzie sprawdzać obowiązującą obecnie teorię we współczesnej kardiologii o tym, co stanowi przyczynę występowania miażdżycy i w efekcie powodowania chorób układu krążenia. Na potrzeby tego artykułu przyjmiemy sobie, że jest ona hipotezą. Będzie ona dotyczyć cholesterolu, który znajduje się w układzie krążenia. To automatycznie oznacza, że nie będziemy mówić ani o cholesterolu w diecie, ani o cholesterolu w organizmie ogółem. Powiązanie cholesterolu w diecie z cholesterolem w układzie krążenia to temat na osobne opracowanie. Natomiast cholesterol jako substancja budulcowa błon komórkowych (oraz niezbędna do tworzenia wielu kluczowych hormonów i związków w organizmie) jest też czymś zupełnie innym niż cholesterol w układzie krążenia. My skupiamy się w tekście tylko na tym ostatnim (a nawet jeszcze bardziej wąsko). Nie ma on tak naprawdę istotnego związku z tym, jak cholesterol jest syntetyzowany w innych tkankach organizmu (np. w mózgu, który jest odcięty od dostępu do cholesterolu we krwi).
W rzeczywistości dla wielu zaskoczeniem może się okazać, że większość cholesterolu w organizmie znajduje się w tkankach obwodowych. Zaś tego interesującego najbardziej, czyli zawartego w tak zwanych „lipoproteinach” w układzie krążenia jest mniej niż 10 procent. Nawet czerwone krwinki mają go więcej zgodnie z tabelą 1 (Hellerstein & Turner, 2014):

Tabela 1. Zawartość cholesterolu w organizmie (źródło Hellerstein et al. 2014)
Lipidogram i lipoproteiny, czyli o pasażerach i transporterach
Zacznijmy od tak zwanego lipidogramu, który jest podstawą dla kardiologów do oceny ryzyk chorób serca. Pojawiają się na nim cztery wielkości: całkowity cholesterol, cholesterol HDL, cholesterol LDL i trójglicerydy. Ich zależność między sobą jest zazwyczaj następująca przy założeniu, że stosujemy jednostki pomiaru stosowane zazwyczaj w Polsce (czyli miligramy na decylitr):
Całkowity cholesterol = (cholesterol HDL) + (cholesterol LDL) + (trójglicerydy/5)
To równanie już na wstępie wprowadza trochę zamieszania. Cholesterol to cholesterol, jedna i ta sama substancja. Może się znajdować w różnych miejscach, więc taki rozdział może mieć sens, ale co tam nagle robią trójglicerydy? Wszak to inna substancja niż cholesterol. Z jakiej racji ma wobec tego sumować się do całkowitej puli?
Otóż równanie jest szacowaniem. Zazwyczaj u większości ludzi sprawa wygląda tak, że jak od całkowitego cholesterolu odejmiemy cholesterol HDL oraz cholesterol LDL, to zostaje taki cholesterol resztkowy, który statystycznie rzecz biorąc jest w ilości pięciokrotnie niższej niż trójglicerydy (przy jednostkach mg/dL). Dlatego równanie tak naprawdę wygląda tak:
Całkowity cholesterol = (cholesterol HDL) + (cholesterol LDL) + (cholesterol w reszcie)
A tak się składa, że „cholesterol w reszcie” to statystycznie zazwyczaj pięć razy mniej w miligramach na decylitr niż trójglicerydów (żeby skomplikować sprawę, to w jednostkach innych, stosowanych np. w USA przelicznik jest inny). Niestety nie jest to ścisła zależność i zdarzają się pacjenci, którzy (zwłaszcza przy wysokich trójglicerydach) nie zachowują takiej liczbowej zależności. Jest to o tyle niewygodne, że większość laboratoriów nie mierzy bezpośrednio cholesterolu LDL (chociaż są takie metody), tylko mierzy go właśnie w oparciu o to równanie.
Zostawmy na razie to zagmatwanie i spróbujmy ogarnąć sprawę samego transportu cholesterolu we krwi. To będzie centralne do zrozumienia mechanizmów fizjologicznych: ani cholesterol, ani trójglicerydy nie mogą przemieszczać się same po krwiobiegu. Wie o tym każda osoba, która kiedykolwiek wlała do szklanki z wodą olej: to się nie miesza. Dlatego w celu przemieszczania po krwiobiegu niezbędne są odpowiednie „łodzie podwodne”, które nazywamy lipoproteinami. Jak sama nazwa wskazuje są to cząsteczki, które zawierają w sobie różne tłuszcze, szeroko rozumiane lipidy (cholesterol, trójglicerydy itd.), oraz proteiny czyli po prostu białka. Ponieważ białko lubi wodę, to stanowi doskonały budulec do wykonania łodzi podwodnej dla cholesterolu i tłuszczu, transportowanego po organizmie.
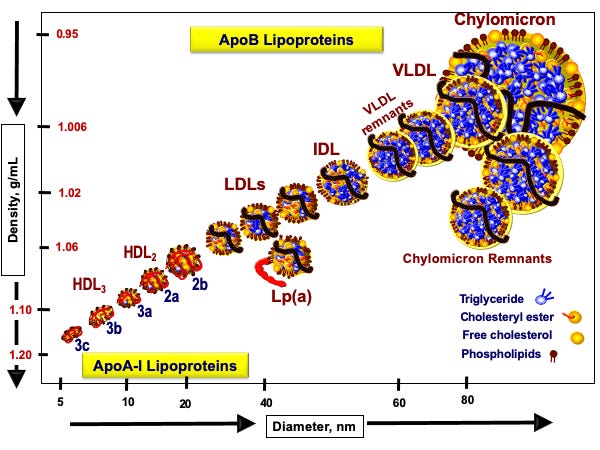
A zatem przyjrzymy się rysunkowi 1 (Nakajima, 2014), by zobaczyć, jak te pojazdy lipoproteinowe wyglądają:
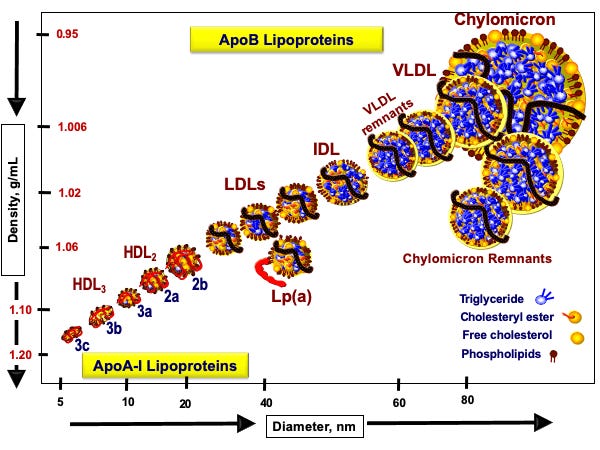
Rysunek 1. Klasy lipoprotein na podstawie Nakajama 2014 i strony www.peterattia.com
Oś dolna pokazuje średnicę naszej łodzi podwodnej (lipoproteiny). Oś boczna pokazuje gęstość łodzi podwodnej. Im łódź podwodna jest mniejsza, tym po prostu ma większą gęstość, jest poniekąd bardziej „napakowana”. Dlatego najmniejsze i najbardziej gęste, jak sama nazwa wskazuje, są lipoproteiny HDL, a więc High Density Lipoproteins. Niedaleko od nich są lipoproteiny większe jak sama nazwa wskazuje o mniejszej gęstości, czyli Low Density Lipoproteins. Wśród nich jest jedna złośliwa lipoproteina Lp(a), którą warto raz w życiu sobie przebadać.
Następnie mamy lipoproteiny trochę większe od LDL, które nazywamy lipoproteinami o średniej gęstości, Intermediate Density Lipoproteins, a następnie lipoproteiny jeszcze większe o bardzo małej gęstości, Very Low Density Lipoproteins. Generalnie piszę trochę tautologicznie, ponieważ im większa lipoproteina, tym mniejszą ma gęstość. Liczba tych lipoprotein generalnie porusza się w przeciwnym kierunku do wielkości. Najwięcej liczbowo mamy cząstek HDL, a z pośród tych nie-HDL najwięcej mamy cząstek LDL.
Na rysunku cholesterol i jego estry jest zaznaczony na żółto, a trójglicerydy na niebiesko. Widzimy, że jako pasażerowie jedne i drugie pakują się w różnych ilościach do różnych pojazdów. Pojazdy te odróżniamy nie tylko wielkością, ale przede wszystkim typem białek, które się w nich znajdują. Jak widzimy pojazdy HDL mają czerwoną tablicę rejestracyjną. To oznacza, że ich kursy są realizowane do wątroby. Natomiast cząsteczki z czarną tablicą rejestracyjną, cząsteczki nie-HDL (LDL, IDL, VLDL) realizują inne kursy.
W tym miejscu dochodzimy do kluczowego sedna hipotezy lipidowej we współczesnej kardiologii. Pojazdy z czarnymi tablicami rejestracyjnymi, które mają mniejszą średnicę niż 70 nanometrów, są w stanie wchodzi do ścian tętnic. Dzieje się tak za sprawą ich czarnej tablicy rejestracyjnej, a konkretnie za sprawą tak zwanego białka apoB. Jeśli cząstka takie białko zawiera, to swoisty szlaban w ścianie tętnicy może się podnieść i taką cząstkę (przewożącą cholesterol i trójglicerydy) wpuścić.
Nota bene istnieją proste i standaryzowane testy laboratoryjne, które sprawdzają liczbę cząstek apoB. Ponieważ każda z nich jest jednym pojazdem, to zawiera tylko jedną tablicę rejestracyjną. Mierząc zatem stężenie apoB w organizmie dokonuje się poniekąd pomiaru liczby tych cząstek, zdolnych do przedostawania się do ścian tętnic. To dosyć istotny szczegół, który warto pamiętać w trakcie czytania tego tekstu.
W zasadzie zamiast mówić o „cholesterolu HDL”, „cholesterolu nie-HDL” i „cholesterolu LDL” dużo lepiej byłoby mówić o „cholesterolu w HDL”, „cholesterolu w nie-HDL” i „cholesterolu w LDL”. Cholesterol jest jeden i ten sam, tylko podróżuje w różnych pojazdach. W praktyce klinicznej zatem mierzy się następujące rzeczy:
· „Całkowity cholesterol” – cały cholesterol podróżujący we wszystkich lipoproteinach (ale nie cały we krwi, bo więcej jego jest np. w krwinkach, ani tym bardziej nie jest to cały cholesterol w organizmie).
· „Cholesterol HDL” – cały cholesterol podróżujący w cząstkach HDL.
· „Cholesterol nie-HDL” – cały cholesterol podróżujący w lipoproteinach z białkiem apoB (zazwyczaj cholesterol w nie-HDL koreluje się z liczbą apoB w krwiobiegu, ale nie w 100 procentach).
· „Cholesterol LDL” – cały cholesterol podróżujący w lipoproteinach LDL (można zrobić osobny test sprawdzający liczbę cząstek LDL, który jest skorelowany, ale nie w 100 procentach, z samą ilością zawartego w nich cholesterolu).
· „Trójglicerydy” – tłuszcze podróżujące we wszystkich lipoproteinach, przy czym w głównej mierze zazwyczaj podróżują one w większych cząstkach apoB.
2. Hipoteza lipidowa w kardiologii, czyli skąd rzekomo bierze się miażdżyca i w konsekwencji choroby układu krążenia?
W pewnym sensie już widzimy, dlaczego postawione w tytule pytanie jest nieścisłe. Gdyby traktować je literalnie, to odpowiedź na nie brzmi po prostu „nie”. Współczesna kardiologia zresztą nie twierdzi, że procesy niszczące układ krążenia wynikają z cholesterolu we krwi. Każdy mechanizm opisowy, który znajdziemy w publikacjach mówi o konkretnych lipoproteinach, czyli naszych łodziach podwodnych. Nie zaś o ich pasażerach. Dlaczego zatem ciągle medycyna posługuje się lipidogramem i stężeniami cholesterolu oraz trójglicerydów? To pytanie pewnie wymagające osobnej analizy. My zatrzymajmy się w tym miejscu na europejskich wytycznych, wskazanych przez dwie prestiżowe organizacje, European Society of Cardiology oraz European Atherosclerosis Society, które sprawę stawiają dosyć jednoznacznie (Mach et al., 2020):

Rysunek 2. Wytyczne europejskie dotyczące lipidów (Mach et al., 2020)
„Biorąc pod uwagę główną przyczynową rolę lipoprotein zawierających apoB w inicjacji i progresji miażdżycy, bezpośredni pomiar krążącego stężenia aterogennych lipoprotein zawierających ApoB byłby idealnym rozwiązaniem w celu zarówno oszacowania ryzyka, jak i ukierunkowania decyzji dotyczących leczenia.”
Nie jest to zresztą tylko stanowisko dwóch prestiżowych organizacji. Kiedy sięgniemy po publikacje naukowe opisujące rzekomy proces miażdżycy, to jako siłę napędową przedstawia się właśnie transportery, konkretne lipoproteiny, a nie to, co przewożą (Borén et al., 2020; Tabas et al., 2007; Williams & Tabas, 1995). W sumie nic dziwnego, ponieważ liczy się pojazd wjeżdżający w ścianę tętnicy, a nie to, jakich dokładnie przewozi pasażerów.
W zasadzie zatem nasza hipoteza robocza może już na starcie zostać zmodyfikowana. Pytanie nie brzmi, czy cholesterol w układzie krążenia powoduje choroby układu krążenia (pomijając już, że jest go dużo w krwinkach czerwonych). Pytanie brzmi: czy lipoproteiny apoB (transportujące cholesterol i trójglicerydy) powodują choroby układu krążenia? Do lipoprotein apoB zaliczają się cząstki LDL (główny składnik), IDL i VLDL.
Jedno, co bez wątpienia możemy powiedzieć o miażdżycy, to to, że jest chorobą akumulacyjną. Nie rozwija się tak szybko jak choroby wirusowe. Rozwija się nawet nie latami, a dekadami życia. Dokonując obrazowania tętnic ludzi w różnym wieku można dostrzec różne stadia tej choroby, pokazujące, że następuje ona w powolny, ale systematyczny sposób, co widać na rysunku 3 (Nakashima et al., 2007):
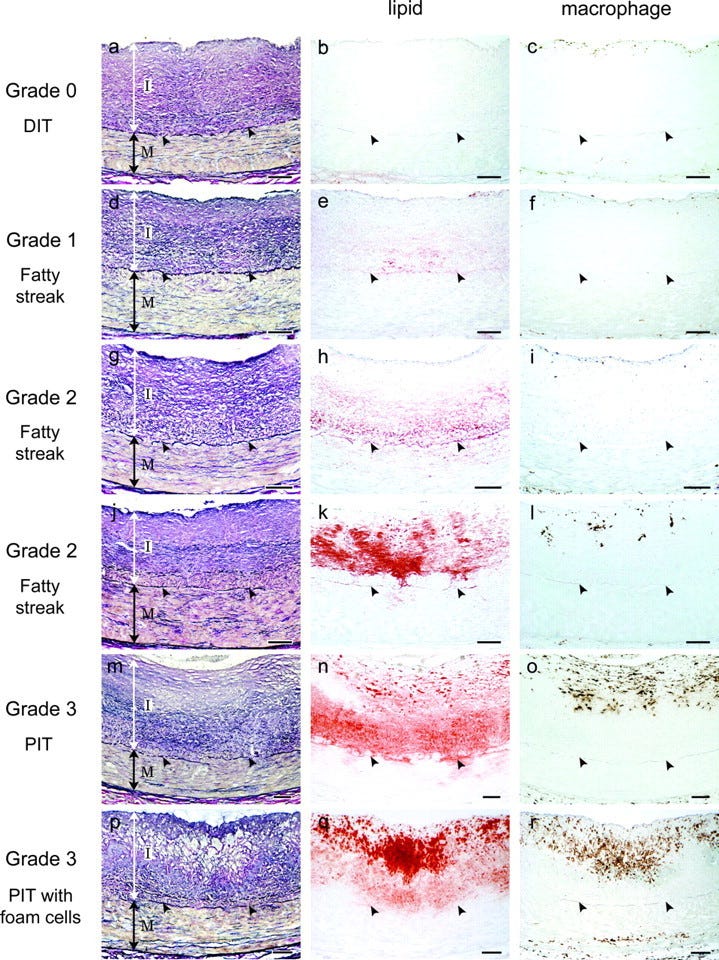
Rysunek 3. Akumulacja procesów miażdżycowych (Nakashima et al. 2007)
Początki miażdżycy zresztą obserwuje się u bardzo młodych ludzi, którym robiono autopsję, a którzy np. zginęli w wyniku jakiegoś wypadku (Joseph et al., 1993). Nie jest to również cecha charakterystyczna dla rozwoju cywilizacyjnego, ponieważ miażdżycę odkrywa się również w zmumifikowanych ciałach (Allam et al., 2011). Jest więc coś immanentnego w organizmach ludzkich, co czyni je podatne na powolne, ale konsekwentne niszczenie tętnic. Innymi słowy, ponieważ choroby układu krążenia to główny zabójca kobiet i mężczyzn, to człowiek pracuje na niego przez całe życie dokładnie tak samo jak pracuje całe życie na emeryturę. Jeśli ktoś chce wpłynąć na swoją sytuację materialną na starość, to musi zacząć myśleć o oszczędzaniu po wejściu w dorosłość. Nie inaczej jest z miażdżycą, gdzie działa zasada procentu składanego: jeśli ktoś chce uniknąć na starość zawału i udaru, to musi zacząć redukować swoje ryzyka już w młodości.
Czołowa teza współczesnej kardiologii na temat patologii miażdżycy brzmi następująco: duża i długa ekspozycja na podniesioną liczbę lipoprotein apoB transportujących cholesterol i trójglicerydy powoduje miażdżycę, a w konsekwencji choroby układu krążenia. Proces ten jest opisywany w następujący sposób: cząstki apoB przedostają się do ściany tętnic, gdzie w przypadku części z nich następuje ich retencja, a więc zatrzymanie. W konsekwencji uruchamiać to ma reakcję zapalną i powolną budowę „blaszek” miażdżycowych, które przez lata ulegają następnie zwapnieniu (choć nie zawsze). Zmniejszać to ma przepływ krwi, co sprawiać ma w przyszłości potencjalne problemy albo z powodu mniejszego dopływu lub nagłego odklejenia się stwardniałej blaszki miażdżycowej. Główna linia procesu przedstawia zatem następującą kolejność: więcej cząstek apoB —> większa penetracja śródbłonka —> więcej retencji —> więcej procesów aterogennych i stanu zapalnego (proszę zwrócić uwagę, że zgodnie z tą hipotezą zapalenie jest konsekwencją już uruchomionych procesów, a nie pierwszym i niezbędnym czynnikiem zapalnym: do ścian tętnic wchodzą lipoproteiny nieutlenione).
W praktyce wiele danych populacyjnych testujących tę hipotezę opiera się nie na pomiarze apoB wprost, lecz tego co w wysokim stopniu z nim koreluje, czyli stężeniu cholesterolu w LDL, lub cholesterolu nie-HDL.
Miażdżyca jest chorobą wieloczynnikową. Gdybyśmy mogli w oparciu o tezy kardiologiczne sformułować jej model do sfalsyfikowania, wzór na nią, to wyglądałby tak:
Miażdżyca = F (liczba lipoprotein apoB, nadciśnienie, palenie, cukrzyca, inne stany zapalne, procesy naturalnego rozpadu, Y) x czas
Miażdżyca miałaby być wobec tego funkcją liczby cząstek apoB, gdyż te traktuje się jako niezbędne do wprowadzenia cholesterolu i lipidów do ścian tętnic. Większa liczba cząstek z czarnymi tablicami rejestracyjnymi by po prostu zwiększała szansę na to, że dojdzie do ich retencji i w konsekwencji uruchomienia stanu zapalnego, produkującego blaszkę miażdżycową. Jednocześnie sama liczba cząstek apoB, choć miałaby być czynnikiem koniecznym, to mogłaby być przyspieszana lub spowalniana. Nadciśnienie, palenie papierosów, wysokie poziomy glukozy we krwi (cukrzyca), wszelkie stany zapalne oraz naturalne procesy rozpadu w organizmie będą ten proces zdecydowanie przyspieszać. W nawiasie zawarłem też Y jako być może jakieś czynniki, które w przyszłości zostaną jeszcze odkryte.
Wszystkie te czynniki mnożymy przez czas, ponieważ gra on kluczową rolę. Przy sprawdzaniu ryzyka zawału czy udaru, pierwsza najważniejsza informacja o pacjencie dotyczy jego/jej wieku. Ale wiek to tylko liczba, jak mawiał klasyk. Wiek jest traktowany w ten sposób nie dlatego, że to jakaś zmienna fundamentalna. Wiek jest traktowany jako symbol akumulacji wszystkich negatywnych czynników z przeszłości. Nie jest ważne palenie papierosów w tej chwili, tylko jak długo się je pali. Nie jest ważne, czy naturalne procesy rozpadu w tętnicach obecnie trwają, tylko jak długo już trwają. Nie jest ważne, czy w danej chwili ma się nadciśnienie, tylko jak długo się żyje z tym nadciśnieniem. Nie jest ważny podniesiony poziom glukozy, lecz jak długo go mamy podniesionego. I wreszcie w rozumowaniu kardiologicznym nie jest ważny pomiar apoB w danym momencie, lecz jak długo dany poziom jest utrzymywany. To dlatego „wiek” jest najważniejszą zmienną w ryzyku: ponieważ symbolizuje wszystko, co się dotychczas zakumulowało. A miażdżyca to – jak już mówiliśmy – przede wszystkim choroba akumulacyjna.
Nie jest mi dane rozstrzygać, na ile te procesy fizjologiczne mają sens. Nie jestem lipidologiem, nie jestem patologiem, nie jestem fizjologiem. Jednakże tak jak pisaliśmy już we wpisie o cukrzycy, mechanistyczne spekulacje nie są tak naprawdę priorytetowe w medycynie i zdrowiu, gdyż ostatecznie o sile danej hipotezy/teorii decyduje zgodność z danymi populacyjnymi. I to właśnie zamierzamy sprawdzić.
Jak sfalsyfikować hipotezę kardiologii?
Tak jak zaznaczyłem przed chwilą, nie bardzo mam jakiekolwiek pole do tego, aby wnikać w przedstawione przez współczesną kardiologię mechanizmy miażdżycy, nawet jeśli dobrze współgrają z wszelkimi badaniami laboratoryjnymi na zwierzętach. Możemy jednak skonstruować na podstawie tej teorii przewidywanie. Jeśli prawdziwa jest hipoteza o tym, że długa i duża ekspozycja na cząstki apoB transportujące cholesterol podnosi ryzyka sercowo-naczyniowe, to u ludzi z takim czynnikiem ryzyka powinno występować statystycznie więcej udarów, zawałów i innych incydentów sercowo-naczyniowych.
Schemat postępowania i potencjalnej falsyfikacji wygląda zgodnie z rysunkiem 4 poniżej:

Rysunek 4. Próba falsyfikowania hipotezy o roli danego czynnika ryzyka (schemat własny)
Postępowanie wygląda tak, że w danej populacji staramy się stworzyć dwie bardzo podobne do siebie, niemalże identyczne grupy. Zwracamy uwagę na to, aby obydwie grupy miały podobne wszelkie istotne charakterystyki: wiek, płeć, BMI, jednostki chorobowe, ciśnienie krwi itd. Grupy różnicujemy tylko i wyłącznie pod jednym kątem: interesującego nas czynnika ryzyka. Jedna grupa ma być poddana w większym stopniu ekspozycji na cząstki podejrzane jako miażdżycogenne (z czarnymi tablicami rejestracyjnymi), a druga grupa będzie się od niej różnić tym, że będzie poddana tym cząstkom w mniejszym stopniu.
Tego typu eksperyment można przeprowadzać na trzy zupełnie niezależne sposoby, stanowiące osobne kategorie dowodowe i osobne źródło wiedzy. Każdy z tych trzech sposobów będzie przez nas uwzględniony. Hierarchia dowodzenia w naukach medycznych wygląda z grubsza tak jak na rysunku 5 (Davies et al., 2018):

Rysunek 5. Hierarchia dowodzenia w naukach medycznych (Davies et al. 2018)
Na samym szczycie jakości i mocy dowodzenia znajdują się randomizowane badania kliniczne RCT (randomized clinical trials), czyli takie, gdzie ludzie projektują badanie poprzez tworzenie dwóch homogenicznych grup pod każdym możliwym względem (nad nimi są systematyczne przeglądy takich badań). Następnie jedna z grup jest poddana interwencji medycznej, a druga otrzymuje placebo. Próba jest podwójnie ślepa, czyli ani pacjenci nie wiedzą, kto otrzymuje placebo, ani bezpośrednio medycy, którzy się nimi zajmują.
Następne w hierarchii będą tak zwane Mendlowskie randomizacje i badania kohortowe, które omówię niżej.
3. Próba falsyfikacji
Weryfikacja bezpośrednia czyli co mówią badania kliniczne?
Kiedy dokonujemy oceny materiału dowodowego, to zawsze należy brać pod uwagę nie jedno wybrane badanie, ale tak dużo badań jak to tylko możliwe. Tak, by poszerzyć spektrum dowodowe, a nie skupić się tylko na jakimś jednym (np. małym lub krótkim) badaniu klinicznym. Ponieważ kluczowy jest całościowy obraz sytuacji, to należy starać się spojrzeć na dany problem z możliwie najszerszej perspektywy. To samo dotyczy badań klinicznych i na szczęście mamy dobre narzędzie statystyczne, które do tego służy: meta-analiza. Spójrzmy zatem na meta-analizę 26 niezależnych od siebie badań klinicznych farmakoterapii obniżających cholesterol w LDL, przeprowadzonych w sumie na 170 tysiącach pacjentów. Tak zwane drzewko meta-analizy opublikowanej w prestiżowym The Lancet przedstawione jest na rysunku 6 (“Efficacy and Safety of More Intensive Lowering of LDL Cholesterol: A Meta-Analysis of Data from 170 000 Participants in 26 Randomised Trials,” 2010):

Rysunek 6. Drzewko meta-analizy efektów 26 badań farmakoterapii obniżającej cholesterol w LDL
Pierwsze pięć badań na drzewku to porównanie małej dawki statyn do większej dawki. Kolejne 21 badań to porównanie statyn do grupy placebo. Każdy wiersz to osobne badanie. Prawie wszystkie pokazują „wychylenie” w lewą stronę, co oznacza, że przyjmowanie statyn miało tendencję do tego, aby obniżać ryzyka wystąpienia incydentów sercowo-naczyniowych (zawały, udary itd.). Niektóre z tych badań nie osiągnęły tak zwanej „statystycznej istotności”, ale na szczęście meta-analiza pozwala je razem zsumować. Efektem jest wiersz „subtotal”, który reprezentowany jest diamencikiem pokazującym, jak bardzo te interwencje zmniejszały ryzyko. W sumie uśredniając interwencja obniżająca cholesterol LDL o około 39 mg/dL skutkowała zmniejszeniem liczby występowanych incydentów o 22 procent.
W tej samej meta-analizie wprowadzono też pogrupowanie pacjentów pod kątem rozmaitych czynników ryzyka, żeby sprawdzić, czy nie jest tak, że statyny działają na przykład tylko na kobiety, albo tylko na cukrzyków, albo tylko na ludzi o niskim HDL itd. Analiza podgrup jest przedstawiona na rysunku 7:

Rysunek 7. Drzewko meta-analizy wprowadzające podział pacjentów na określone grupy ryzyka
Praktycznie żaden osobny czynnik ryzyka nie wpływał na efekty oddziaływania redukcji cholesterolu w LDL w organizmie. Bez względu na to, czy się pali, czy nie, czy się ma otyłość, nadwagę, czy normalną masę ciała, czy się ma nadciśnienie, czy go nie ma, czy się jest mężczyzną, czy kobietą, czy się ma historię już chorób serca, czy nie, czy ma się wysoki cholesterol w HDL, czy niski, czy jest się starszym, czy młodszym – efekt oddziaływania statyn jest ten sam przez cały przekrój badanych. Sugeruje to bardzo mocno, że mamy do czynienia z silnym i całkowicie niezależnym od reszty czynnikiem ryzyka incydentów sercowo-naczyniowych.
W ostatnim roku ukazała się inna meta-analiza badań klinicznych, która z kolei pogrupowała badania pod kątem długości ich trwania. Pamiętamy, że hipoteza lipoproteinowa mówi nie tylko o wielkości ekspozycji na większe bądź mniejsze poziomy aterogennych cząstek, ale także o upływie czasu na tę ekspozycję. Wyniki są przedstawione na rysunku 8 (N. Wang et al., 2022):

Rysunek 8. Drzewko meta-analizy pokazujące efekty badań klinicznych na redukcję ryzyka w zależności od długości trwania terapii (N. Wang et al. 2022)
Z opracowania wynika, że tak jak w świecie finansów istnieje procent składany, to samo dotyczy systematycznej redukcji ryzyka w wyniku obniżenia cholesterolu w LDL w organizmie. Plansza A pokazuje efekt po roku terapii i choć wychylenie jest widoczne, to jest niewielkie. Czerwony diament uśredniający wszystkie niezależnie badania coś pokazuje, ale w niewielkim zakresie. Kolejna plansza B już pokazuje trochę większe efekty po 2 latach. C po 3 latach i tak dalej. Stopniowo skumulowany efekt jest coraz większy i istotniejszy. Największy efekt oczywiście widać po siedmiu latach, choć z tego powodu, że badań siedmioletnich było mniej, to jest mniejsza pewność statystyczna tego efektu. W sumie jednak widać tu wyraźnie efekt składanej procentowej redukcji ryzyka w wyniku stosowanej farmakoterapii. Im dłużej cholesterol w LDL pozostawał niższy, tym większe benefity w zmniejszaniu incydentów. Przeprowadzona z badań tak zwana meta-regresja jest przedstawiona na rysunku 9:
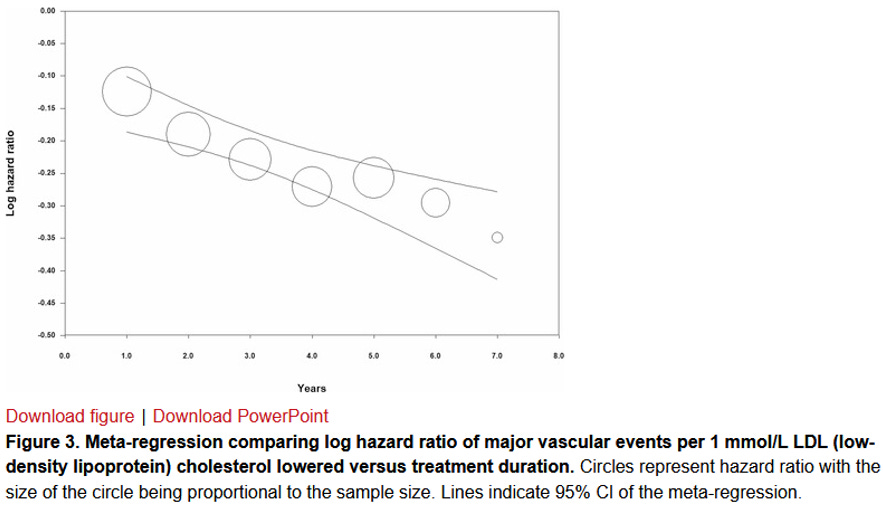
Rysunek 9. Meta-regresja pokazująca, że wydłużanie czasu trwania terapii obniżającej cholesterol w LDL skutkuje kumulacyjnym efektem w postaci redukowania ryzyka incydentów sercowo-naczyniowych (N. Wang et al. 2022)
Generalnie wykres oddaje efekt kumulacyjny oddziaływania poprzez farmakoterapię i przez to redukowania udarów, zawałów i innych negatywnych zdarzeń sercowo-naczyniowych.
Inna meta-analiza statyn z roku 2022 pokazywała to samo zjawisko: że znaczenie ma nie tylko sama wielkość obniżonego cholesterolu w LDL w organizmie, ale również okres trwania, przez jaki to obniżenie następowało. W sumie stworzono analizę na aż ćwierć milionie ludzi (co jest liczbą ogromną w przypadku badań klinicznych) z 33 randomizowanych badań. Wnioski są takie, że zarówno intensywność terapii jak i czas trwania mają ogromne znaczenie (Redel-Traub et al., 2022).
Badania obserwacyjne, kohortowe, czyli dłuższe, ale mniej dokładne
Postępowanie dowodowe nie kończy się tylko na badaniach klinicznych. Ekspozycja na dany czynnik ryzyka występuje przecież w populacji naturalnie. I analogicznie do badania klinicznego można prześledzić zdrowotnie ludzi, którzy mają podobne do siebie czynniki ryzyka, ale różnią się tym jednym, który nas interesuje. Te badania mają pewną istotną słabość wobec badań klinicznych, ale jednocześnie i pewną przewagę, jeśli chodzi o naszą hipotezę. Z jednej strony, w ich przypadku słabiej się kontroluje czynniki zakłócające badanie. Z drugiej strony, są przeprowadzone nie w horyzoncie cztero- pięcioletnim, ale przez lat kilkanaście. Można zatem zaobserwować rzekome działanie czynnika ryzyka w dłuższym okresie.
Jedna z takich meta-analiz kohort w populacji na ponad milionie uczestników porównywała grupy ludzi o różnych poziomach cholesterolu w LDL w trzech przedziałach: (1) mniej niż 70 mg/dL, (2) 70-130 mg/dL, oraz (3) więcej niż 130 mg/dL. Porównanie najwyższego przedziału do reszty pokazywało wzrost śmiertelności i ryzyka sercowo-naczyniowego. W przypadku niższych przedziałów widać było podobne „wychylenia”, ale nie osiągnięto statystycznej istotności (Peng et al., 2022).
Inna meta-analiza z kolei zgromadziła najistotniejsze i szczególnie znane kohorty (między innymi Health Professionals, Nurses’ Health, Framingham Offspring, INTERHEART, Women’s Health) by sprawdzić, jak zmiany w lipoproteinach podnoszą ryzyka sercowo-naczyniowe. Co ciekawe, analiza dokonała tak zwanego przeglądu „niezgodności”, gdyż sprawdzała, który marker naczyniowy jest skuteczniejszy w przewidywaniu ryzyka. I chociaż wpływ LDL-C (cholesterol w LDL) był istotny klinicznie i statystycznie, to z jednej strony przebijał go pomiar nie-HDL-C, ale najlepiej ryzyko ważył wskaźnik liczby cząstek, czyli opisany przez nas wcześniej apoB. Pokazuje to rysunek 10 (Sniderman et al., 2011):

Rysunek 10. Analiza „niezgodności”, która pokazuje, że w najistotniejszych kohortach w pomiarze markerów ryzyka sercowo-naczyniowego wskaźnik cholesterolu w nie-HDL jest lepszy od wskaźnika cholesterolu w LDL. Jednocześnie oba wskaźniki wypadają gorzej niż apoB (Sniderman et al. 2011)
„Eksperymenty Pana Boga” („Eksperymenty Matki Natury”) czyli mendlowskie randomizacje (MR)
Na koniec omawianej hipotezy przejdziemy do specyficznego materiału dowodowego, który z jednej strony wyrasta z koncepcji badań kohortowych, ale z drugiej strony jest zbliżony do badań klinicznych. Chodzi o tak zwane mendlowskie randomizacje. Są to genetyczne kohorty, ale w przeciwieństwie do klasycznych kohort nie zawierają w sobie problemu czynników zakłócających. Nie są to jednocześnie badania kliniczne, gdyż na pewno wszystko nie jest tak samo kontrolowane, ale z drugiej strony mają horyzont czasowy nie do pobicia, gdyż rozciągają się na całe życie pacjenta.
W mendlowskiej randomizacji badacze poszukują pojedynczej mutacji genetycznej, która odpowiada za jedną istotną zmianę w organizmie człowieka. Każdy z nas może mieć np. losową mutację, która podnosi stężenie metabolitów witaminy D w organizmie. Inni mogą takowej nie mieć. To oznacza, że w populacji tworzy się w randomizowany sposób losowa grupa, która ma zmieniony jeden czynnik ryzyka. Następnie wystarczy tę podgrupę prześledzić (np. pod kątem incydencji nowotworów), by się przekonać, czy podnoszenie (lub obniżenie) witaminy D w organizmie przyczynia się do zmniejszenia lub zwiększenia występowania raka. Nazwać to można „eksperymentem Pana Boga” lub „eksperymentem Matki Natury” (jak kto woli), ponieważ jest to losowe badanie, trochę udające badanie kliniczne. Jednocześnie, chociaż trochę słabiej kontroluje czynniki zakłócające, to ma dwie zasadnicze przewagi nad badaniami typowo klinicznymi. Po pierwsze, MR przeprowadzone są na dużo większej próbce populacyjnej. Po drugie, są przeprowadzone na dużo dłuższym horyzoncie czasowym, kilkudziesięciu lat, gdyż przez całe życie. To sprawia, że w niektórych kwestiach mendlowskie randomizacje dają nawet większą pewność dowodową aniżeli badania kliniczne.
Jeśli chodzi o mutacje genetyczne, to jest wiele różnych typów, które obniżają poziomy zawartości lipoprotein z apoB w organizmie. Rysunek 11 zbiera te różne typy mutacji w zależności od wielkości średniego obniżenia stężenia apoB i zestawia to z redukcją ryzyka incydentów sercowo-naczyniowych (Ference et al., 2019):

Rysunek 11. Zależność genetycznego obniżenia stężenia cząstek apoB i redukcji ryzyka chorób układu krążenia (Ference et al. 2019)
Dwie rzeczy szczególnie są uderzające na tej ilustracji. Po pierwsze, liniowość zależności. Rzadko kiedy w naukach medycznych zależność od dawki układa się niemalże w linię prostą, wskazując na istnienie twardej i konsekwentnej zależności. Po drugie, efekt jest widoczny nawet przy niewielkich obniżkach stężenia apoB. W badaniach klinicznych takich liczb byśmy nie zobaczyli, gdyż trwają one powiedzmy pięć lat. Miażdżyca natomiast rozwija się przez trzydzieści, czterdzieści i więcej lat. Mutacja genetyczna z kolei obniża stężenie cząstek apoB nie przez pięć lat, lecz przez całe życie, od urodzenia, czyli jeszcze zanim miażdżyca zacznie się rozwijać. Dlatego nawet mniejsza redukcja dawki powoduje swoistego rodzaju leczniczy efekt akumulacyjny. Analogiczny efekt zobaczymy oszczędzając małe sumy pieniędzy przez 40 lat, a potem porównując to z oszczędzaniem przez 10 lat.
Wcześniejsza meta-analiza obejmująca mniej mutacji pokazywała taką samą zaskakującą liniowość. Jednocześnie warto ją przytoczyć, ponieważ zestawiono w niej w pary „eksperymenty Pana Boga” („Matki Natury”) z eksperymentami człowieka, czyli mendlowskie randomizacje z badaniami klinicznymi. Pogrupowano je w zależności od wielkości osiągniętego obniżenia cholesterolu w LDL. Wyniki przedstawia rysunek 12 (Ference et al., 2012):

Rysunek 12. Zestawienie genów i interwencji farmakologicznych, które osiągają taką samą obniżkę cholesterolu w LDL (Ference et al. 2012)
Obniżka o jakąś ilość cholesterolu w LDL poprzez mutację genetyczną daje zawsze większe redukcje ryzyka sercowo-naczyniowego aniżeli taka sama obniżka przez interwencję farmakologiczną. Skąd zatem różnica? Jak już kilka razy podkreślaliśmy, liczy się nie tylko wielkość obniżki, lecz także czas przez jaki oddziałuje. Geny ma się od urodzenia, pigułki od czasu wypisania recepty. Obniżenie cholesterolu w LDL o około 39 mg/dL przez okres pięciu lat obniża ryzyka zawałów i udarów w zasadzie o około 24 procent (trochę za mało miejsca mam tu by wskazać obliczenia, ale to bez znaczenia dla sedna argumentu). Takie samo obniżenie za pomocą genów powoduje dużo większą redukcję ryzyka, bo zdecydowanie ponad 50 procent (też daruję obliczenia). Analogiczne różnice dostrzeżemy przy porównywaniu do siebie niższych obniżek. Jest to zgodne z akumulacyjną koncepcją powstawania miażdżycy. I także zgodne z główną hipotezą kardiologiczną.
Kompilacja wszystkich dowodów na raz: genetycznych, interwencyjnych i obserwacyjnych
Badania obserwacyjne z kohort, mendlowskie randomizacje, badania interwencyjne kliniczne – wszystkie te analizy stosują odmienne od siebie metody. Stanowią od siebie niezależne źródła wiedzy. Wszystkie te trzy grupy badań można wobec tego zestawić ze sobą w całość, by stworzyć jedno wielkie spektrum dowodowe, tak jak uczyniono to w słynnym raporcie z roku 2017 (Ference et al., 2017). Najważniejszy wykres z tego opracowania znajduje się na rysunku 13:
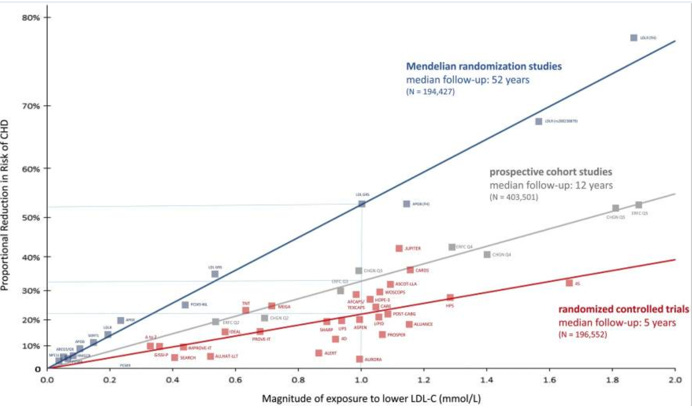
Rysunek 13. Kompilacja badań klinicznych, obserwacyjnych i genetycznych obrazująca wpływ obniżki cholesterolu w LDL na ryzyko występowania incydentów sercowo-naczyniowych (Ference et al. 2017)
Raczej rzadko w naukach medycznych i o zdrowiu zdarza się, by zależność była tak jednoznaczna i jednocześnie spójna czasowo. Poszczególne kwadraty na wykresie to pojedyncze badania. Czerwonym kolorem zaznaczone są badania kliniczne randomizowane. Czerwona krzywa pokazuje uśredniony efekt redukcji cholesterolu w LDL na ryzyka w przypadku tych badań. Szary kolor symbolizuje badania kohortowe. Szara krzywa pokazuje już mocniejsze konsekwencje (uśrednione dla kohort) ze względu na to, że obszar badań dotyczy średnio 12 lat, a nie 5 jak w przypadku badań klinicznych. Jest to w pełni zgodne z akumulacyjnym efektem wpisanym w hipotezę lipidową.
Najbardziej jednak efekt akumulacyjny widać w przypadku badań genetycznych (niebieskie kwadraty). Dla porównania na wykresie łatwo odczytać uśredniony efekt czasowy np. patrząc na obniżkę cholesterolu w LDL o 1 mmol/L, czyli około 39 mg/dL. Badania kliniczne (czas około 5 lat) powodują redukcję ryzyka średnio o około 22 procent. W kohortach (średnio 12 lat) powoduje to już redukcję o 33 procent. Natomiast w mendlowskich randomizacjach (średnio 52 lata) redukcja osiąga już ponad 50 procent. W jednym z badań na czarnej amerykańskiej populacji mutacja w PCSK9 bardzo radykalnie obniżająca cholesterol w LDL (o 28 procent) doprowadzała nawet do szokującej redukcji ryzyka sercowo-naczyniowego aż o 88 procent (ponad ośmiokrotna redukcja zawałów i udarów!) (Cohen et al., 2006).
W tym miejscu wspomnę jeszcze jedną meta-analizę, która grupowała razem rozmaite sposoby obniżania stężenia cholesterolu w LDL. W sumie sposobów na osiągnięcie tego jest kilka różnych poprzez różne szlaki i to nie tylko za pomocą statyn. Są oprócz tego inhibitory PCSK9, operacje jelitowe, leki wiążące kwasy żółciowe, interwencje dietetyczne, fibraty, niacyna, ezetymib, inhibitory CETP. W sumie mamy 9 różnych niezależnych sposób obniżania cholesterolu w LDL w organizmie, a ich efekt został naniesiony na rysunku 14 (Silverman et al., 2016):
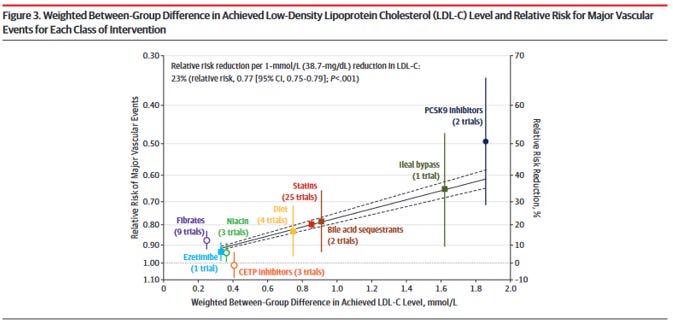
Rysunek 14. Redukcja cholesterolu w LDL za pomocą różnych metod a redukcja poważnych incydentów sercowo-naczyniowych (Silverman et al. 2016)
Praktycznie wszystkie interwencje – poza jedną – wyznaczają analogiczny kierunek, odpowiadający „dawce”. Im bardziej obniżany jest cholesterol w LDL, obojętnie którym szlakiem i jaką metodą, tym wyraźniejszy efekt redukcji ryzyka sercowo-naczyniowego. Spośród dziewięciu metod tylko jedna nie przyniosła takiego rezultatu: inhibitory CETP, ale akurat wiemy dlaczego… ponieważ sztucznie podnosiły cholesterol w HDL, co okazało się błędem, gdyż cholesterol w HDL bywa niezłym markerem na poziomie populacji, ale najprawdopodobniej nie jest wprost przyczynowo powiązany z chorobami układu krążenia (lecz jedynie korelacyjnie na poziomie populacji).
Możemy zatem uznać, że próba falsyfikacji hipotezy lipidowej kończy się porażką. Zależność od wielkości dawki (redukcji lub podwyższenia cząstek aterogennych) oraz od długości ekspozycji na tę dawkę jest niezwykle silnie potwierdzona wieloma rozmaitymi badaniami na milionach ludzi i na przestrzeni kilkudziesięciu lat. Trudno znaleźć w medycynie lepiej skoroborowaną (filozoficzne określenie na „potwierdzoną”) hipotezę niż ta.
Teraz możemy przejść do kilku zastrzeżeń powiązanych z tą hipotezą, które przewijają się nie tyle w prasie naukowej, ile głównie w mediach społecznościowych.
4. Pewne zarzuty do teorii kardiologicznej
„To nie cholesterol w LDL, tylko stan zapalny powoduje miażdżycę, LDL to policjant reagujący na przestępstwo”
Ta hipoteza policyjna dosyć często przewija się w niejednym materiale popularyzatorskim, chociaż nie ma potwierdzenia w publikacjach naukowych. Oczywiście zgodnie z akceptowanym modelem miażdżycy stany zapalne odgrywają ogromną rolę w niszczeniu śródbłonka tętnic i przez to ułatwianiu retencji, a następnie akumulacji destrukcyjnych lipoprotein apoB. Jednakże jak widzieliśmy w całym spektrum dowodowym (kohorty, genetyka, interwencje) sama liczba lipoprotein apoB jest niezależnym czynnikiem ryzyka od pozostałych. Trochę to przypomina liczbę aut na drodze. Oczywiście, że liczba wypadków zależy od tego, jaki jest stan drogi, jej organizacja, pora dnia, pogoda etc. Nie zmienia to faktu, że generalnie im więcej aut na drodze, ceteris paribus, tym większa szansa, że dojdzie do kolizji. Nie inaczej w przypadku cząstek apoB – im więcej ich w krwiobiegu, tym większa statystycznie szansa na patogenną retencję w ścianach tętnic. I to jest szansa niezależna od stanów zapalnych, choć te mogą ją znacząco zwiększać.
Hipotezę zapalną można zresztą łatwo zweryfikować tak samo jak weryfikowaliśmy hipotezę lipidową. Oto i przykładowa kohorta 28 tysięcy amerykańskich kobiet, które podzielono na cztery grupy w zależności od markera stanu zapalnego i stężenia cholesterolu w LDL (Ridker et al., 2002). Wyniki obrazuje rysunek 15:

Rysunek 15. Krzywe przeżywalności pacjentek w zależności od poziomów markera stanu zapalnego i cholesterolu w LDL (Ridker et al. 2002)
Wykres przedstawia tak zwane krzywe przeżywalności bez incydentów, czyli odsetek populacji, która nie doświadczyła negatywnych zdarzeń sercowo-naczyniowych. Najbardziej stroma krzywa, szybko opadająca w dół pokazuje, że w tej grupie najwięcej kobiet doświadczyło znaczącego incydentu sercowo-naczyniowego. Jest to krzywa pacjentek o wysokim wyniku CRP (marker stanu zapalnego) oraz wysokim stężeniu cholesterolu w LDL. Z kolei krzywa najbardziej płaska, czyli oznaczająca najzdrowszą część (z najmniejszą liczbą zdarzeń), to grupa kobiet o niskich CRP i niskim LDL-C. Pozostałe dwie grupy to wysokie CPR i niskie LDL-C oraz wysokie LDL-C i niskie CRP. Jak widać, obydwie grupy mają podniesione ryzyka sercowo naczyniowe. Nawet grupa z niskim markerem stanu zapalnego ma podniesione ryzyka, jeśli cholesterol w LDL jest podniesiony. I w sumie nic dziwnego, ponieważ jest to w pełni zgodne z hipotezą lipidową, mówiącą o apoB jako niezależnym od innych czynniku ryzyka (abstrahując nawet od tego, że do badania można wysunąć istotne zastrzeżenie, czy uwzględniono wszystkie czynniki zakłócające). Wykres ma dwa rysunki, gdyż jeden dotyczy całej kohorty, a drugi wyodrębnionej podgrupy pacjentek, które nie stosowały zastępczej terapii hormonalnej. To rozróżnienie pozostaje bez znaczenia dla sedna kwestii, gdyż obie ilustracje pokazują w sumie to samo, choć trochę inne liczby.
„LDL pełni funkcje odpornościowe”
Kolejna teza, która się czasami przewija w przypadku lipoprotein LDL, to sugestia również nawiązującą do retoryki „policjanta z odznaką LDL”, że te cząstki są istotne w funkcjonowaniu układu odpornościowego. Cóż, tak jak pisałem, nie jestem fizjologiem, więc nie mnie oceniać te spekulacje mechanistyczne. Przede wszystkim nawet jeśli ta teza jest prawdziwa, to jeszcze nie oznacza, że im wyższe poziomy cząstek LDL w organizmie, tym lepiej dla układu odpornościowego. Przykładowo glukoza jest absolutnie kluczowa do funkcjonowania mózgu i czerwonych krwinek w organizmie, bez których byśmy poumierali. Glukoza to życie, bez niej czeka nas śmierć. To jednak nie oznacza, że wyższe poziomy glukozy we krwi są niezbędne do tego, by lepiej funkcjonował mózg i krwinki. Dlatego sama z siebie mechanistyczna spekulacja jakoby LDL był potrzebny do sprawnego układu odpornościowego jeszcze nic nie znaczy. Możemy za to spróbować sfalsyfikować tezę, jakoby wyższe poziomy LDL miały sprzyjać zmniejszaniu redukcji infekcji.
I tak w tej kwestii mamy wysyp badań w populacji. Nie ma sensu, żebym przytaczał je wszystkie szczegółowo, więc ograniczę się do kilku przykładów. Trzy meta-analizy kohort pokazywały, że stosowanie statyn redukowało ryzyko śmiertelności na covid-19 (Kollias et al., 2021; Wu et al., 2021; Zein et al., 2022). Inna z kolei meta-regresja nie odnalazła takiej zależności (Hariyanto & Kurniawan, 2021).
Jeden z tak zwanych przeglądów parasolowych (umbrella review), czyli takich, które przeglądają inne systematyczne przeglądy, dotyczący konkretnie stosowania statyn i ryzyka infekcji, doszedł do wniosku, że dowody słabej do przeciętnej jakości wskazują na to, że statyny redukują śmiertelność z powodu infekcji (Ghayda et al., 2021). Jeśli zatem mielibyśmy wyciągać jakieś wnioski, to raczej takie, że albo statyny nie mają wpływu na infekcje i powikłania, albo mają lekki pozytywny wpływ. Jeśli ktoś bardzo chce, to niech sam przejrzy sobie listę 16 meta-analiz dotyczących infekcji płucnych, pooperacyjnych, HIV, czy bakteryjnych, które zamieszczam w przypisie (Chen et al., 2018; Chopra et al., 2012; Deshpande et al., 2015; Duan et al., 2020; Falagas et al., 2008; Janda et al., 2010; Khan et al., 2013; Kwok et al., 2012; B.-X. Ma et al., 2015; Y. Ma et al., 2012; Pertzov et al., 2019; Tleyjeh, 2009; Uthman et al., 2018; Wan et al., 2014; Wijarnpreecha et al., 2019; Zhang et al., 2017).
A jeśli ktoś nie chce tracić na to czasu, to zachowawcze wnioski są takie: meta-analizy kohort i badań klinicznych pokazują albo brak jakiegokolwiek wpływu statyn na infekcję, albo pokazują efekt pozytywny. Tak, czy inaczej, brak jakichkolwiek dowodów na to, by farmakoterapia obniżająca stężenie cząstek apob prowadziła do wzrostu ryzyk infekcji.
To samo zresztą dotyczy ryzyk nowotworowych, ale o nich trochę więcej powiem w sekcji poniżej, gdyż nieskorygowane badania populacyjne wskazują na korelację między niższym całkowitym cholesterolem w lipoproteinach na starość a występowaniem nowotworów.
„Większość ludzi po zawałach ma normalne LDL”
Faktycznie większość ludzi po zawałach może mieć tak zwane „normalne” poziomy LDL w organizmie, ale to nie oznacza falsyfikacji hipotezy lipidowej. Przede wszystkim taka jednostkowa obserwacja nie koliduje w żaden sposób z główną tezą. Pomiar jednostkowy lipidogramu zaraz po zawale nie weryfikuje przecież prawdziwości tezy o akumulacyjnym lipoproteinowym charakterze miażdżycy. Według tej tezy podwyższone poziomy lipoprotein dekadami prowadzą do patologii w układzie krążenia. Teza ta nie mówi, że w momencie zawału lipidogram będzie mieć ten, czy inny poziom, lecz że w przeszłości przez lata lipidogram miał ten, a nie inny poziom.
Statystyczny rozkład poziomów LDL zaraz po zawale zależy od wielu czynników. Przede wszystkim warto wspomnieć o tym, że zaraz po zawale liczba cząstek LDL ma tendencję do tymczasowego obniżania się, nawet o średnio 17 procent (Kumar et al., 2019; Rott et al., 2015). Po drugie, pacjenci z zawałami mogą być już w trakcie terapii obniżającej stężenie lipoprotein w organizmie ze względu na wcześniej zidentyfikowane ryzyka. Jak wiemy już dobrze z efektu akumulacyjnego, taka terapia nie jest w stanie – nawet po kilku latach stosowania – wyeliminować wcześniej zakumulowanego ryzyka. Może raczej tak naprawdę przeciwdziałać temu, żeby to ryzyko się dodatkowo dalej nie podnosiło. Większość ludzi umiera w szpitalach, ale to nie znaczy, że szpitale to miejsca masowych mordów. Szpitale redukują ryzyka zgonu, ale nie anulują ryzyka zakumulowanego wcześniej.
Ponadto sama ta obserwacja („większość ludzi po zawale ma podniesione LDL”) świetnie nadaje się na jedne z pierwszych zajęć ze statystyki, żeby zrozumieć rolę efektu bazowego. Posłużmy się tutaj innym przykładem: większość ludzi, która umiera, nie ma otyłości trzeciego stopnia (BMI powyżej 40). Czy to oznacza, że otyłość trzeciego stopnia nie zwiększa ryzyka zgonu? Oczywiście, że zwiększa i to znacząco, ale ponieważ w populacji takich ludzi jest po prostu mniej, to absolutna liczba zgonów takich osób jest mniejsza niż reszty populacji. Nadal jednak BMI powyżej 40 znacząco podnosi ryzyko ogólnej śmiertelności w stosunku do reszty. Analogicznie: większość ludzi po zawale to nie są palacze papierosów (albo jeszcze lepiej: większość ludzi zaraz po zawale nie pali papierosa…). Większość ludzi na oddziałach nowotworowych to nie są palacze, ale palenie i tak zwiększa ryzyko kluczowych nowotworów. Rozumiemy zależność: kluczowy jest punkt wyjścia, gdy mówimy o rozkładach pewnych efektów.
Ponadto, skoro już jesteśmy przy tak zwanych „normach LDL”, to należy pamiętać, że są one tworzone w oparciu o rozkład populacyjny. To, co bywa nazywane „normą”, może być raczej normą statystyczną niż medycznym zaleceniem. Tak zwane „normalne” poziomy LDL są kompatybilne z rozwojem miażdżycy, co pokazało jedno z badań na rysunku 16 (Fernández-Friera et al., 2017):

Rysunek 16. Zależność cholesterolu w LDL i występowania miażdżycy u pacjentów bez innych czynników ryzyka (Fernández-Friera et al., 2017)
W badaniu przyglądnięto się około 1800 pacjentom z jednej z kohort, z których wyselekcjonowano grupę 740 bez rozpoznanych innych czynników ryzyka sercowo-naczyniowego. Następnie poddano ich badaniom obrazowym, by stwierdzić ewentualne istnienie blaszek miażdżycowych w różnych tętnicach. Wyniki uporządkowano zgodnie z przedziałami zmierzonego poziomu cholesterolu w LDL w organizmie. Wyniki okazały się ponownie dosyć uderzające. Jedynie bardzo niskie poziomy cholesterolu w LDL związane były z bardzo niską szansą na rozwój miażdżycy. Tymczasem to, co bywa nazywane „normą” tego markera było kompatybilne z 40-procentową szansą na powolne akumulowanie w sobie chorób sercowo-naczyniowych (co warte odnotowania: przy średniej wieku 40-kilka lat).
„Ważniejsze od cholesterolu w LDL są trójglicerydy oraz cholesterol w HDL”
Niejednokrotnie można usłyszeć w mediach społecznościowych, że w sumie cholesterol w LDL nie jest istotny, bo ważniejsze są trójglicerydy i cholesterol w HDL, albo stosunek jednego do drugiego. Nie chcę za bardzo tutaj wnikać w szczegóły tych tez, ponieważ trójglicerydy i HDL doczekają się na blogu prawdopodobnie osobnego i szerokiego opracowania, więc na razie skupię się na skrótowym przedstawieniu sytuacji.
Po pierwsze, stosunek trójglicerydów do HDL jest często markerem insulinooporności i ukrytych niebezpieczeństw związanych z ryzykiem cukrzycy. To oznacza, że jest on dobrym markerem, ale niekoniecznie markerem przyczynowym.
Po drugie, co już wspominaliśmy wielokrotnie, miażdżyca to choroba wieloczynnikowa. To, że jeden z czynników niesie ze sobą ryzyka nie wyklucza przecież, że niosą je ze sobą inne.
Po trzecie, gdy mówimy o trójglicerydach, to możemy wrócić do naszej pierwszej sekcji i rysunku 1:
Rysunek 17. Powtórzony rysunek 1
Trójglicerydy nie mogą – podobnie jak cholesterol – samodzielnie podróżować po krwiobiegu. Potrzebują do tego transporterów z czarnymi tablicami, czyli cząstek apoB, które są wielką rodziną pojazdów przenoszących zarówno tłuszcz jak i cholesterol. W tej chwili dyskutowane w literaturze jest, czy trójglicerydy w ogóle są niezależnym od liczby apoB czynnikiem ryzyka sercowo-naczyniowego. Wspomniane przeze mnie wytyczne europejskie tak stawiają sprawę (Mach et al., 2020):
Podwyższone stężenie trójglicerydów (TG) w osoczu wiąże się ze zwiększonym ryzykiem sercowo-naczyniowym, ale związek ten znika po skorygowaniu o stężenia cholesterol nie-HDL, czyli o szacunkowe całkowite stężenie wszystkich lipoprotein zawierających apoB. Podobnie obniżenie stężenia TG za pomocą fibratów zmniejsza ryzyko zdarzeń naczyniowych o taką samą wartość, jak terapie obniżające stężenie cholesterolu LDL-C, w przeliczeniu na jednostkę zmiany stężenia cholesterolu nie-HDL, co sugeruje, że wpływ stężenia trójglicerydów w osoczu na ryzyka jest pośredni poprzez zmiany stężenia lipoprotein bogatych w trójglicerydy, szacowanych na podstawie nie-HDL-C.
Badania z randomizacją mendlowską również sugerują, że związek między stężeniem TG w osoczu a ryzykiem może być przyczynowy; jednak dowody te należy interpretować z ostrożnością, ponieważ prawie wszystkie warianty genetyczne związane z TG są również związane z HDL-C, LDL-C lub Lp(a). W niedawnym badaniu z randomizacją mendlowską wykazano, że warianty obniżające stężenie TG w lipazie lipoproteinowej (LPL) i warianty obniżające stężenie LDL w receptorze LDL miały taki sam wpływ na ryzyko na jednostkę zmiany cząstek ApoB, co sugeruje, że wszystkie lipoproteiny zawierające ApoB mają taki sam wpływ na ryzyko sercowo-naczyniowe. Łącznie badania te silnie sugerują, że przyczynowy wpływ lipoprotein bogatych w trójglicerydy i ich pozostałości na ryzyko zależy od stężenia krążących cząstek zawierających ApoB, a nie od samej zawartości trójglicerydów.
Innymi słowy, według najnowszych wytycznych europejskich trójglicerydy nie wydają się nieść ze sobą ryzyka w oderwaniu od liczby cząstek apoB, które je transportują. Liczy się łódź podwodna, a nie ilość tego, co przewozi. Trochę tak samo było kiedyś z cholesterolem w LDL. Kiedy w badaniach populacyjnych zestawia się stężenie cholesterolu w LDL razem z liczbą cząstek LDL, to okazuje się, że w większości przypadków te zmienne się pokrywają. Kiedy jednak występuje między nimi różnica, to ryzyko wyraźnie podąża za liczbą cząstek LDL, a nie tym, co przewożą (ta sama zasada wychodzi w zestawianiu cholesterolu w nie-HDL z liczbą cząstek apob). Słynna kohorta Framingham Offspring dokładnie to pokazywała: ryzyko podążało za liczbą cząstek LDL, a nie za zawartym w nich cholesterolem (Cromwell et al., 2007). Co jest logiczne z hipotezą lipidową: w ściany tętnic nie wchodzą pasażerowie pojazdów, tylko pojazdy. Cząstka apoB ma potencjał retencyjno-aterogenny niezależnie od tego, czy coś tam „siedzi z tyłu”.
Wygląda na to, że ta sama zasada dotyczy trójglicerydów, które mogą być tylko pasażerami. Generalnie im więcej trójglicerydów (pasażerów), tym zazwyczaj więcej pojazdów z czarną tablicą rejestracyjną (cząstek apoB), które je przewożą. Ale wcale tak być nie musi. Jeśli trójglicerydy rosną, a nie rośnie liczba cząstek apoB, to znaczy, że więcej trójglicerydów jest upakowanych w takiej samej liczbie potencjalnie szkodliwych transporterów. Czy to może podnosić samo w sobie ryzyka? Na to przesądzających dowodów na razie brak. Wiem, że pewnie nie raz słyszeliście o tym, jak to „trójglicerydy są ważniejsze od cholesterolu”, ale nie ma na to dowodów (zresztą sama teza bez konkretnych przedziałów liczb jest pusta).
Nota bene nie wiem też za bardzo jak zwolennicy tezy „trójglicerydy ważniejsze niż LDL” chcą argumentować za swoją teorią w oderwaniu od koncepcji lipoprotein, które je przewożą.
Z cholesterolem zawartym w HDL natomiast sprawa jest również interesująca, ponieważ obserwacyjnie faktycznie da się zauważyć w populacji, że generalnie wyższe stężenia cholesterolu w HDL (tak do górnej granicy 60-70 mg/dL) wydają się być kardioprotekcyjne i wiązać się z mniejszymi ryzykami. Jednocześnie te kohortowe obserwacje stoją w sprzeczności z randomizacjami mendlowskimi i badaniami klinicznymi – oba te typy badań kwestionują, że generalnie wyższy cholesterol w HDL oznacza lepszą sytuację. Zostawię to kiedyś na szersze opracowanie, a tutaj zatrzymajmy się jedynie na tym, że ponownie przytoczyłem w tej kwestii opinię z europejskich wytycznych (Mach et al., 2020).
„Ważne są tylko małe cząstki LDL, a nie duże”
Kolejna z tez potencjalnie zanadto modyfikujących hipotezę lipidową mówi o tym, że cząstki LDL są „heterogeniczne” i nie niosą ze sobą takiego samego ryzyka. Rozwijając,: te mniejsze cząstki LDL miałyby być autentycznie aterogenne w odróżnieniu od tych większych cząsteczek LDL.
Różnicowanie poszczególnych lipoprotein względem siebie, jeśli chodzi o ich ryzyko, nie jest oczywiście niczym nowym ani radykalnym. Jest to podejście wpisane w kardiologię, ponieważ jedną z tych lipoprotein, którą uważa się za szczególnie niebezpieczną, jest cząstka Lp(a). Równie dobrze mogłoby tak być np. z mniejszymi cząstkami LDL w porównaniu do większych cząstek LDL. W tym miejscu jednakże trzeba zaznaczyć dwie bardzo ważne rzeczy. Po pierwsze, naukowcy, którzy wskazują na szkodliwość małych cząstek LDL nie mówią, że tylko małe LDL są aterogenne, lecz, że małe LDL są bardziej aterogenne od większych LDL. Jednym z takich naukowców jest między innymi Ronald Krauss, który od wielu lat propaguje taką wizję (Krauss, 2022). Ten sam naukowiec, który jest współautorem meta-analizy sugerującej, że tłuszcz nasycony niekoniecznie powoduje wzrost ryzyka-sercowo naczyniowego (Siri-Tarino et al., 2010). Przytaczam tę informację celowo, ponieważ Krauss jest jednocześnie współautorem kluczowego opracowania wspomnianego przeze mnie wyżej o wiele mówiącym tytule: Low-density lipoproteins cause atherosclerotic cardiovascular disease. 1. Evidence from genetic, epidemiologic, and clinical studies. A consensus statement from the European Atherosclerosis Society Consensus Panel (Ference et al., 2017). Czyli w skrócie “LDL powoduje miażdżycę”. Tak więc jednoznacznie kluczowy naukowiec promujący tezę o większej aterogenności małych LDL jednocześnie nie przeczy, że wszelkie LDL jest aterogenne.
Pozwolę sobie też w tym miejscu zwrócić uwagę, dlaczego teza o rzekomo większej aterogenności małych cząstek LDL nie jest powszechnie uznawana w kardiologii (ja nie jestem kompetentny do zajęcia w tej sprawie stanowiska, więc staram się przytaczać obecny stan wiedzy). Po pierwsze, małe cząstki LDL to zazwyczaj – obok wspomnianych wysokich trójglicerydów i niskiego HDL – marker stanu cukrzycowego lub przedcukrzycowego. Nie wiadomo wobec tego, do jakiego stopnia byłaby to przyczynowa relacja, a do jakiego po prostu znowu bycie markerem.
Po drugie, badania pokazujące większą szkodliwość małych cząstek LDL do dużych cząstek LDL zazwyczaj nie biorą pod uwagę, że małe i duże cząstki LDL są ze sobą wzajemnie skorelowane. Przy danym stężeniu cholesterolu w LDL w organizmie, większa jego ilość w małych cząstkach LDL od razu oznacza mniejszą ilość w cząstkach dużych LDL. Trochę to zagmatwane, więc skorzystajmy z rysunku 18 (de Graaf et al., 2015):
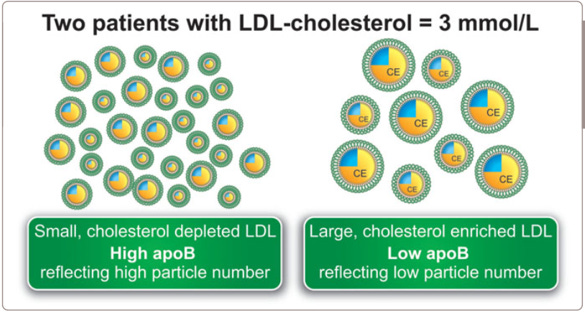
Rysunek 18. Dwóch różnych pacjentów z ta samą ilością cholesterolu w LDL, a jednocześnie zupełnie inną liczbą cząstek apoB i przez to innym ryzykiem (de Graaf et al. 2015)
Rysunek przedstawia dwóch pacjentów o tej samej zawartości cholesterolu w LDL w organizmie. Jednakże ta ilość cholesterolu u jednego pacjenta jest rozbita na więcej mniejszych pojazdów z czarnymi tablicami (cząstek apoB), a w przypadku drugiego pacjenta na znacznie mniej pojazdów z czarnymi tablicami, ale większych. Z powyższego opisu wiemy, że liczba cząstek jest istotniejsza niż sama zawartość cholesterolu. Jest więc jasne, że pacjent z mniejszą liczbą cząstek będzie mieć mniejsze ryzyko niż pacjent z większą liczbą, nawet jeśli sumaryczna zawartość cholesterolu jest taka sama. Sęk w tym, że badacz, który pomija kwestię liczby cząstek i skupia się tylko na rozróżnieniu na małe i duże, dojdzie do wniosku, że przy stałej ilości cholesterolu w LDL w organizmie, małe cząstki będą dużo bardziej aterogenne niż duże cząstki. Przy stałej ilości cholesterolu w tych lipoproteinach większa liczba cząstek niemalże automatycznie pokaże większą aterogenność, bo więcej takich cząstek to analogicznie mniej cząstek dużych. Nie jest do końca jasne, czy skorygowanie o liczbę cząstek apoB wyeliminuje w całości rozróżnienie na małe i duże cząstki LDL. Nauka pewnie przyniesie nam więcej informacji w przyszłości. Warto jednak podkreślić, że ta kwestia została rozpoznana na przykład w bardzo dobrej analizie rodzajów cząstek w kohorcie MESA w roku 2007 (Mora et al., 2007). Słowami autorów: W niektórych badaniach stwierdzono, że mały rozmiar LDL był związany ze zwiększonym ryzykiem sercowym, podczas gdy inne nie potwierdziły niezależności tego związku. W dwóch małych badaniach (…) nie stwierdzono niezależnego związku z pomiarami wielkości cząstek LDL. Inne badania sugerowały, że duży rozmiar LDL może być związany z ryzykiem naczyniowym. Jednak większość raportów odnoszących się do wielkości LDL badała tylko dystrybucję podklas LDL lub fenotyp wielkości LDL (duży lub mały), a nie stężenia podklas LDL. Stwierdziliśmy, że aterogenność dużych LDL stała się widoczna tylko wtedy, gdy osoby zostały sklasyfikowane według niskiego lub wysokiego poziomu małych LDL lub po uwzględnieniu poziomów małych LDL za pomocą korekty statystycznej.
„Niski cholesterol zwiększa ogólną śmiertelność”
Na sam koniec czeka nas najdłuższa analiza, ale analiza konieczna ze względu na powszechność zarzutu: w populacji często obserwuje się, szczególnie u starszych ludzi, zależność niższego cholesterolu ze zwiększoną śmiertelnością. Prym na przykład wiedzie odnaleziony w czeluściach mediów społecznościowych następujący rysunek 19:

Rysunek 19. Zbierając pasywnie dane statystyczne wychodzi na to, że ludzie z niskim cholesterolem więcej umierają
Zanim wyjaśnimy dokładniej powyższe zjawisko, to musimy sobie coś ustalić, co prawdopodobnie powinienem być ustalić bardziej szczegółowo przed napisaniem tego tekstu. Postępowanie w nauce wymaga zastosowania odpowiednich metod dowodzenia. Takie metody dowodzenia i hierarchie są obecne w naukach o zdrowiu i medycznych. To oznacza, że nie każda publikacja, nie każdy zestaw danych i nie każdy wykres mają taką samą wagę. W przeciwnym razie wpadniemy w wir chaosu i kompletnego niezrozumienia tego, jak dokonuje się rzeczywista i konkretna analiza.
Wyobraźmy sobie sytuację, w której przed sądem stoi osoba oskarżona o morderstwo. W jej sprawie mamy następujące dowody potwierdzające jej niewinność: badanie DNA na miejscu zbrodni, mocne alibi potwierdzone przez kilkunastu świadków, oraz nagrania z kamer osiedlowych. Jednocześnie oprócz tego pojawia się starsza ledwo poruszająca się i półślepa emerytka, mieszkająca kilkadziesiąt metrów od miejsca zbrodni, która twierdzi, że widziała tę osobę jednak na miejscu zbrodni w momencie jej popełnienia. Co w takiej sytuacji zrobi sąd? Mimo iż zdecyduje się na poświęcenie jej uwagi, to w ostatecznym rozrachunku zignoruje jej zeznania, gdyż nagrania cyfrowe, badania genetyczne oraz zeznania kilkunastu innych niezależnych świadków będą potraktowane jako priorytetowe wobec osoby, której obserwacyjne zdolności są mocno ograniczone.
Z tym samym mamy do czynienia w tej sytuacji z rysunku 19. Przypomnijmy hierarchię dowodzenia na rysunku 20:

Rysunek 20. Hierarchia dowodzenia w medycynie pokazująca, że waga badań kohortowych, randomizowanych i mendlowskich jest dużo większa od prostego badania obserwacyjnego.
W istocie rysunek 19 klasyfikuje się jako tak zwane badanie „ekologiczne”, czyli dokonujące pasywnej obserwacji dla całych grup. Tego typu badania są obarczone ogromnym ryzykiem błędów z powodu niemożliwości kontrolowania wielu zakłócających czynników. W istocie rysunek 19 doskonale nadaje się na jedne z pierwszych zajęć na przedmiocie „epidemiologia”, by pokazać studentom niebezpieczeństwa opierania się na tego typu obserwacjach. Otwórzmy polski podręcznik do epidemiologii, który w trochę trudnych słowach (zaraz je objaśnimy) opisuje pułapki kryjące się w badaniu „ekologicznym”. Profesor Jan Zejda pisze w ten sposób (Paradowska-Stankiewicz I et al., 2021):
Historia epidemiologii, nie wykluczając tej najnowszej, zawiera szereg przykładów pułapki ekologicznej definiowanej jako skutek milczącego założenia, że zależności przyczynowo-skutkowe obserwowane na poziomie populacyjnym/grupowym odzwierciedlają zależności obecne na poziomie indywidualnym… Wśród innych okoliczności o niekorzystnym wpływie na interpretację wyników badania ekologicznego można wymienić:
· Brak możliwości kontroli czynników zakłócających
· Heterogenność grup w zakresie wielkości narażenia i/lub występowania zjawiska zdrowotnego
· Brak czułości wobec następstwa czasowego (narażenie a efekt zdrowotny)
· Zniekształcający wyniki efekt współliniowości związany ze zjawiskiem korelacji różnych czynników traktowanych jako potencjalne czynniki ryzyka
W największym uproszczeniu przy tego typu prostych obserwacjach nie sposób odnaleźć twardych zależności przyczynowych, gdyż wiele czynników zależy od siebie nawzajem lub ma wspólne przyczyny (np. w zimie jest więcej stłuczek i grzania kaloryferami, ale jedno nie powoduje drugiego). Szczególnie, jeśli nie tylko patrzymy na całe grupy społeczności, ale kiedy porównujemy ze sobą tak różne społeczności. Na wykresie 19 mamy przecież zaznaczone całe kraje np. Niemcy i Burkina Faso. Z pewnością te kraje różnią się nie tylko średnimi poziomami cholesterolu, ale wieloma innymi czynnikami wpływającymi na śmiertelność.
Dlatego niemalże z automatu wszelkie badania „ekologiczne” przegrywają z badaniami mendlowskimi, klinicznymi i kohortowymi (czyli takimi, które przytoczyliśmy wcześniej).
Jeśli niższe poziomy cholesterolu miałyby zwiększać ogólną śmiertelność, to meta-analizy statyn powinny pokazywać jej wzrosty w przypadku stosowania terapii. Tak nie jest, pokazują rzecz odwrotną (Nowak et al., 2022; Yebyo et al., 2019). Jeśli niższe poziomy cholesterolu w lipoproteinach mają zwiększać ogólną śmiertelność, to szczególnie narażeni powinni być ludzie z mutacjami genetycznymi, które to czynią. Tak nie jest, „eksperymenty Pana Boga” pokazują raczej coś odwrotnego (Benn et al., 2019; Daghlas & Gill, 2021; Postmus et al., 2015). Szczególną uwagę chcę zwrócić na ostatnie badanie z przypisu (Postmus et al., 2015), ponieważ stanowi ono korektę trzech duńskich kohort, w tym kohorty Leiden. Przytaczam ją, ponieważ zdarzyło mi się już na jednej z prezentacji na Youtube widzieć, że referent przytoczył kohortę Leiden pokazującą, że niższy cholesterol wśród starszych ludzi wiąże się ze zwiększoną śmiertelnością. Przy prostej obserwacji tak, ale kiedy dokładnie te same dane skorygowano o cały okres życia (a nie patrzono tylko na jego końcówkę), wyniki wychodzą dokładnie przeciwne: „Wyniki obecnego badania wskazują, że genetyczna predyspozycja do wysokiego poziomu LDL-C przyczynia się do śmiertelności w całym życiu, w tym u najstarszych osób w podeszłym wieku, a korzystny profil ryzyka genetycznego LDL wiąże się z rodzinną długowiecznością”.
Chociaż słabe dowody można odrzucić ze względu na ich ewidentną słabość, to nie zmienia to faktu, że możemy się im przyjrzeć i wyjaśnić, skąd w ogóle taka zależność jak na rysunku 19.
Przede wszystkim wiemy już od dawna, że spadający „całkowity cholesterol” pod koniec życia jest markerem poważnych chorób: niektórych nowotworów, czy poważnych chorób wątroby (Iribarren et al., 1995). Nic więc dziwnego, że mamy do czynienia z odwróconą przyczynowością: to nie niski cholesterol powoduje większą śmiertelność, lecz zbliżająca się wielkimi krokami zwiększona śmiertelność obniża pod koniec życia poziomy cholesterolu. Niski cholesterol w lipoproteinach jest również markerem jeszcze bardziej oczywistego problemu: niedożywienia (Brown & Beyer, 1994; Zarny & Bernstein, 1995). Starsi ludzie często na końcu swojej drogi zaczynają spożywać coraz to mniej jedzenia. Może to być wynikiem problemów gastrycznych, neurologicznych, nowotworowych, psychiatrycznych i wielu innych. W rezultacie następuje przeobrażenie ich lipidogramu. Nie jest to jednak autentyczna medyczna „korekta”, lecz zazwyczaj symptom poważnych problemów. To właśnie dlatego na wynikach kart laboratoryjnych jest „dolna norma” cholesterolu w lipoproteinach. W rzeczywistości kardiologia dzisiaj powie, że z medycznego punktu widzenia takiej dolnej granicy póki co nie ma odkrytej. Zaznacza się ją jednak po to, by lekarze zwrócili uwagę na nagle bardzo niskie poziomy, jeśli nie wynikają one z farmakoterapii lub zmiany stylu życia. Podobnie jeśli bardzo otyła osoba zaczyna nagle mocno chudnąć. Jeśli nie jest to efekt zastosowanego leku lub zmiany stylu życia (diety i ruchu), to najprawdopodobniej mamy do czynienia z ukrytą chorobą.
Na sam koniec przytoczę jeszcze przykład pewnej kohorty chińskiej, składającej się z ponad 41 tysięcy uczestników, gdzie również zademonstrowany był „cholesterolowy paradoks” – ludzie z niższym cholesterolem w nie-HDL więcej umierali, co pokazuje rysunek 21 (B. Wang et al., 2022):
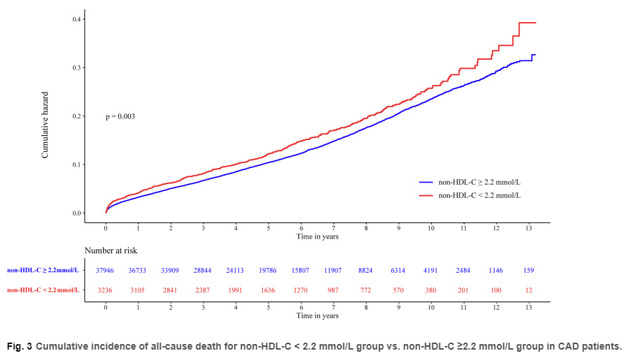
Rysunek 21. Wyższa śmiertelność ludzi o niższym cholesterolu w nie-HDL (czerwona linia) w porównaniu do ludzi o wyższym nie-HDL (niebieska linia) (B. Wang et al. 2022)
Jednakże jak pokazali autorzy, wystarczy kohortę skorygować o pewne czynniki ryzyka, a wtedy okazuje się, że sytuacja się zmieni. Rysunek 22 przedstawia taką korektę:

Rysunek 22. Zmieniające się ryzyko cholesterolu w nie-HDL w zależności od uwzględnienia pozostałych czynników ryzyka (B. Wang et al. 2022).
Pierwsza linia obrazuje wzrost ryzyka zgonu przy niższym cholesterolu w nie-HDL (wychylenie na prawo). Druga linia pokazuje efekt po korekcie o kilka czynników ryzyka. Wychylenie nadal na niekorzyść niskiego nie-HDL, choć już statystycznie nieistotne. Trzecia linia obrazuje, co się dzieje, gdy model uwzględni niedożywienie. Po uwzględnieniu niedożywienia okazuje się, że niższe poziomy cholesterolu nie-HDL obniżają ryzyko zgonu, a nie podwyższają. Czwarty model uwzględnia wszystkie możliwe zaobserwowane w kohorcie czynniki i również pokazuje, że niższy cholesterol w nie-HDL zmniejsza śmiertelność.
W tym miejscu nadmienię, że badacze często mogą wykazywać bardzo dobrą wolę do tego, by swoje modele należycie skorygować o zmienne zakłócające. Niestety nie zawsze jest to możliwe i niestety nie zawsze możemy to zauważyć. Po prostu nie wiemy z dużą dozą pewności, czy dostatecznie wyeliminowaliśmy czynniki zakłócające, czy nie. Dlatego mamy coś takiego jak hierarchię dowodzenia w naukach medycznych. By umieć odsiać ziarno od plew i zauważyć te dowody, które mają zdecydowanie większą moc od pozostałych.
Podsumowanie
Myślę, że jakieś konkretne podsumowanie jest w tym miejscu zbędne. Jeśli ktoś przyjrzał się uważnie przytoczonemu przeze mnie materiałowi dowodowemu i jego jakości, to w tym miejscu nie muszę niczego dodawać.
Jeśli nie, to sugeruję powtórkę lektury.
Literatura:
Allam, A. H., Thompson, R. C., Wann, L. S., Miyamoto, M. I., Nur el-Din, A. el-H., el-Maksoud, G. A., Al-Tohamy Soliman, M., Badr, I., el-Rahman Amer, H. A., Sutherland, M. L., Sutherland, J. D., & Thomas, G. S. (2011). Atherosclerosis in Ancient Egyptian Mummies. JACC: Cardiovascular Imaging, 4(4), 315–327. https://doi.org/10.1016/j.jcmg.2011.02.002
Benn, M., Tybjærg-Hansen, A., & Nordestgaard, B. G. (2019). Low LDL Cholesterol by PCSK9 Variation Reduces Cardiovascular Mortality. Journal of the American College of Cardiology, 73(24), 3102–3114. https://doi.org/10.1016/j.jacc.2019.03.517
Borén, J., Chapman, M. J., Krauss, R. M., Packard, C. J., Bentzon, J. F., Binder, C. J., Daemen, M. J., Demer, L. L., Hegele, R. A., Nicholls, S. J., Nordestgaard, B. G., Watts, G. F., Bruckert, E., Fazio, S., Ference, B. A., Graham, I., Horton, J. D., Landmesser, U., Laufs, U., … Ginsberg, H. N. (2020). Low-density lipoproteins cause atherosclerotic cardiovascular disease: pathophysiological, genetic, and therapeutic insights: a consensus statement from the European Atherosclerosis Society Consensus Panel. European Heart Journal, 41(24), 2313–2330. https://doi.org/10.1093/eurheartj/ehz962
Brown, K. A., & Beyer, P. L. (1994). Low serum cholesterol and risk for protein calorie malnutrition. Journal of the American Dietetic Association, 94(9), A38. https://doi.org/10.1016/0002-8223(94)91711-6
Chen, M., Ji, M., & Si, X. (2018). The effects of statin therapy on mortality in patients with sepsis. Medicine, 97(31), e11578. https://doi.org/10.1097/MD.0000000000011578
Chopra, V., Rogers, M. A. M., Buist, M., Govindan, S., Lindenauer, P. K., Saint, S., & Flanders, S. A. (2012). Is Statin Use Associated with Reduced Mortality After Pneumonia? A Systematic Review and Meta-analysis. The American Journal of Medicine, 125(11), 1111–1123. https://doi.org/10.1016/j.amjmed.2012.04.011
Cohen, J. C., Boerwinkle, E., Mosley, T. H., & Hobbs, H. H. (2006). Sequence Variations in PCSK9, Low LDL, and Protection against Coronary Heart Disease. New England Journal of Medicine, 354(12), 1264–1272. https://doi.org/10.1056/NEJMoa054013
Cromwell, W. C., Otvos, J. D., Keyes, M. J., Pencina, M. J., Sullivan, L., Vasan, R. S., Wilson, P. W. F., & D’Agostino, R. B. (2007). LDL particle number and risk of future cardiovascular disease in the Framingham Offspring Study—Implications for LDL management. Journal of Clinical Lipidology, 1(6), 583–592. https://doi.org/10.1016/j.jacl.2007.10.001
Daghlas, I., & Gill, D. (2021). Low‐density lipoprotein cholesterol and lifespan: A Mendelian randomization study. British Journal of Clinical Pharmacology, 87(10), 3916–3924. https://doi.org/10.1111/bcp.14811
Davies, N. M., Holmes, M. v, & Davey Smith, G. (2018). Reading Mendelian randomisation studies: a guide, glossary, and checklist for clinicians. BMJ, k601. https://doi.org/10.1136/bmj.k601
de Graaf, J., Couture, P., & Sniderman, A. D. (2015). The Life History of ApoB Lipoprotein Particles. In ApoB in Clinical Care (pp. 17–64). Bohn Stafleu van Loghum. https://doi.org/10.1007/978-90-368-0980-1_1
Deshpande, A., Pasupuleti, V., & Rothberg, M. B. (2015). Statin Therapy and Mortality from Sepsis: A Meta-analysis of Randomized Trials. The American Journal of Medicine, 128(4), 410-417.e1. https://doi.org/10.1016/j.amjmed.2014.10.057
Duan, H., Liu, T., Zhang, X., Yu, A., & Cao, Y. (2020). Statin use and risk of tuberculosis: a systemic review of observational studies. International Journal of Infectious Diseases, 93, 168–174. https://doi.org/10.1016/j.ijid.2020.01.036
Efficacy and safety of more intensive lowering of LDL cholesterol: a meta-analysis of data from 170 000 participants in 26 randomised trials. (2010). The Lancet, 376(9753), 1670–1681. https://doi.org/10.1016/S0140-6736(10)61350-5
Falagas, M. E., Makris, G. C., Matthaiou, D. K., & Rafailidis, P. I. (2008). Statins for infection and sepsis: a systematic review of the clinical evidence. Journal of Antimicrobial Chemotherapy, 61(4), 774–785. https://doi.org/10.1093/jac/dkn019
Ference, B. A., Ginsberg, H. N., Graham, I., Ray, K. K., Packard, C. J., Bruckert, E., Hegele, R. A., Krauss, R. M., Raal, F. J., Schunkert, H., Watts, G. F., Borén, J., Fazio, S., Horton, J. D., Masana, L., Nicholls, S. J., Nordestgaard, B. G., van de Sluis, B., Taskinen, M.-R., … Catapano, A. L. (2017). Low-density lipoproteins cause atherosclerotic cardiovascular disease. 1. Evidence from genetic, epidemiologic, and clinical studies. A consensus statement from the European Atherosclerosis Society Consensus Panel. European Heart Journal, 38(32), 2459–2472. https://doi.org/10.1093/eurheartj/ehx144
Ference, B. A., Kastelein, J. J. P., Ray, K. K., Ginsberg, H. N., Chapman, M. J., Packard, C. J., Laufs, U., Oliver-Williams, C., Wood, A. M., Butterworth, A. S., di Angelantonio, E., Danesh, J., Nicholls, S. J., Bhatt, D. L., Sabatine, M. S., & Catapano, A. L. (2019). Association of Triglyceride-Lowering LPL Variants and LDL-C–Lowering LDLR Variants With Risk of Coronary Heart Disease. JAMA, 321(4), 364. https://doi.org/10.1001/jama.2018.20045
Ference, B. A., Yoo, W., Alesh, I., Mahajan, N., Mirowska, K. K., Mewada, A., Kahn, J., Afonso, L., Williams, K. A., & Flack, J. M. (2012). Effect of Long-Term Exposure to Lower Low-Density Lipoprotein Cholesterol Beginning Early in Life on the Risk of Coronary Heart Disease. Journal of the American College of Cardiology, 60(25), 2631–2639. https://doi.org/10.1016/j.jacc.2012.09.017
Fernández-Friera, L., Fuster, V., López-Melgar, B., Oliva, B., García-Ruiz, J. M., Mendiguren, J., Bueno, H., Pocock, S., Ibáñez, B., Fernández-Ortiz, A., & Sanz, J. (2017). Normal LDL-Cholesterol Levels Are Associated With Subclinical Atherosclerosis in the Absence of Risk Factors. Journal of the American College of Cardiology, 70(24), 2979–2991. https://doi.org/10.1016/j.jacc.2017.10.024
Ghayda, R. A., Han, C. H., Lee, K. H., Kim, J. S., Kim, S. E., Hong, S. H., Kim, M., Kronbichler, A., Tizaoui, K., Li, H., Koyanagi, A., Jacob, L., Kim, M. S., Yon, D. K., Lee, S. W., Kostev, K., Shin, J. I., Yang, J. W., & Smith, L. (2021). The effect of statins on mortality among patients with infection: umbrella review of meta-analyses. European Review for Medical and Pharmacological Sciences, 25(6), 2685–2695.
Hariyanto, T. I., & Kurniawan, A. (2021). Statin and outcomes of coronavirus disease 2019 (COVID-19): A systematic review, meta-analysis, and meta-regression. Nutrition, Metabolism and Cardiovascular Diseases, 31(6), 1662–1670. https://doi.org/10.1016/j.numecd.2021.02.020
Hellerstein, M., & Turner, S. (2014). Reverse cholesterol transport fluxes. Current Opinion in Lipidology, 25(1), 40–47. https://doi.org/10.1097/MOL.0000000000000050
Iribarren, C., Reed, D. M., Chen, R., Yano, K., & Dwyer, J. H. (1995). Low Serum Cholesterol and Mortality. Circulation, 92(9), 2396–2403. https://doi.org/10.1161/01.CIR.92.9.2396
Janda, S., Young, A., FitzGerald, J. M., Etminan, M., & Swiston, J. (2010). The effect of statins on mortality from severe infections and sepsis: A systematic review and meta-analysis. Journal of Critical Care, 25(4), 656.e7-656.e22. https://doi.org/10.1016/j.jcrc.2010.02.013
Joseph, A., Ackerman, D., Talley, J. D., Johnstone, J., & Kupersmith, J. (1993). Manifestations of coronary atherosclerosis in young trauma victims—An autopsy study. Journal of the American College of Cardiology, 22(2), 459–467. https://doi.org/10.1016/0735-1097(93)90050-B
Khan, A. R., Riaz, M., bin Abdulhak, A. A., Al-Tannir, M. A., Garbati, M. A., Erwin, P. J., Baddour, L. M., & Tleyjeh, I. M. (2013). The Role of Statins in Prevention and Treatment of Community Acquired Pneumonia: A Systematic Review and Meta-Analysis. PLoS ONE, 8(1), e52929. https://doi.org/10.1371/journal.pone.0052929
Kollias, A., Kyriakoulis, K. G., Kyriakoulis, I. G., Nitsotolis, T., Poulakou, G., Stergiou, G. S., & Syrigos, K. (2021). Statin use and mortality in COVID-19 patients: Updated systematic review and meta-analysis. Atherosclerosis, 330, 114–121. https://doi.org/10.1016/j.atherosclerosis.2021.06.911
Krauss, R. M. (2022). Small dense low-density lipoprotein particles: clinically relevant? Current Opinion in Lipidology, 33(3), 160–166. https://doi.org/10.1097/MOL.0000000000000824
Kumar, N., Kumar, S., Kumar, A., Shakoor, T., & Rizwan, A. (2019). Lipid Profile of Patients with Acute Myocardial Infarction (AMI). Cureus. https://doi.org/10.7759/cureus.4265
Kwok, C. S., Yeong, J. K.-Y., Turner, R. M., Cavallazzi, R., Singh, S., & Loke, Y. K. (2012). Statins and associated risk of pneumonia: a systematic review and meta-analysis of observational studies. European Journal of Clinical Pharmacology, 68(5), 747–755. https://doi.org/10.1007/s00228-011-1159-4
Ma, B.-X., Li, H., Li, J.-S., & Wu, S.-S. (2015). Effect of statins on preventing infectious complications after surgery: Systematic review and meta-analysis. Journal of International Medical Research, 43(5), 610–618. https://doi.org/10.1177/0300060515583708
Ma, Y., Wen, X., Peng, J., Lu, Y., Guo, Z., & Lu, J. (2012). Systematic Review and Meta-Analysis on the Association between Outpatient Statins Use and Infectious Disease-Related Mortality. PLoS ONE, 7(12), e51548. https://doi.org/10.1371/journal.pone.0051548
Mach, F., Baigent, C., Catapano, A. L., Koskinas, K. C., Casula, M., Badimon, L., Chapman, M. J., de Backer, G. G., Delgado, V., Ference, B. A., Graham, I. M., Halliday, A., Landmesser, U., Mihaylova, B., Pedersen, T. R., Riccardi, G., Richter, D. J., Sabatine, M. S., Taskinen, M.-R., … Patel, R. S. (2020). 2019 ESC/EAS Guidelines for the management of dyslipidaemias: lipid modification to reduce cardiovascular risk. European Heart Journal, 41(1), 111–188. https://doi.org/10.1093/eurheartj/ehz455
Mora, S., Szklo, M., Otvos, J. D., Greenland, P., Psaty, B. M., Goff, D. C., O’Leary, D. H., Saad, M. F., Tsai, M. Y., & Sharrett, A. R. (2007). LDL particle subclasses, LDL particle size, and carotid atherosclerosis in the Multi-Ethnic Study of Atherosclerosis (MESA). Atherosclerosis, 192(1), 211–217. https://doi.org/10.1016/j.atherosclerosis.2006.05.007
Nakajima, K. (2014). Remnant Lipoproteins: A Subfraction of Plasma Triglyceride-Rich Lipoproteins Associated with Postprandial Hyperlipidemia. Clinical & Experimental Thrombosis and Hemostasis, 1(2), 45–53. https://doi.org/10.14345/ceth.14013
Nakashima, Y., Fujii, H., Sumiyoshi, S., Wight, T. N., & Sueishi, K. (2007). Early Human Atherosclerosis. Arteriosclerosis, Thrombosis, and Vascular Biology, 27(5), 1159–1165. https://doi.org/10.1161/ATVBAHA.106.134080
Nowak, M. M., Niemczyk, M., Florczyk, M., Kurzyna, M., & Pączek, L. (2022). Effect of Statins on All-Cause Mortality in Adults: A Systematic Review and Meta-Analysis of Propensity Score-Matched Studies. Journal of Clinical Medicine, 11(19), 5643. https://doi.org/10.3390/jcm11195643
Paradowska-Stankiewicz I, Rosińska M, Wojtyniak B, & Zieliński A (Eds.). (2021). Epidemiologia. Od teorii do praktyki. Wydanie 1. PZWL.
Peng, K., Li, X., Wang, Z., Li, M., & Yang, Y. (2022). Association of low-density lipoprotein cholesterol levels with the risk of mortality and cardiovascular events: A meta-analysis of cohort studies with 1,232,694 participants. Medicine, 101(48), e32003. https://doi.org/10.1097/MD.0000000000032003
Pertzov, B., Eliakim-Raz, N., Atamna, H., Trestioreanu, A. Z., Yahav, D., & Leibovici, L. (2019). Hydroxymethylglutaryl-CoA reductase inhibitors (statins) for the treatment of sepsis in adults – A systematic review and meta-analysis. Clinical Microbiology and Infection, 25(3), 280–289. https://doi.org/10.1016/j.cmi.2018.11.003
Postmus, I., Deelen, J., Sedaghat, S., Trompet, S., de Craen, A. J., Heijmans, B. T., Franco, O. H., Hofman, A., Dehghan, A., Slagboom, P. E., Westendorp, R. G., & Jukema, J. W. (2015). LDL cholesterol still a problem in old age? A Mendelian randomization study. International Journal of Epidemiology, 44(2), 604–612. https://doi.org/10.1093/ije/dyv031
Redel-Traub, G., Smilowitz, N. R., Xia, Y., & Berger, J. S. (2022). Systematic review and meta-regression on the duration of LDL-C lowering and major adverse cardiovascular events. Vascular Medicine, 27(4), 375–376. https://doi.org/10.1177/1358863X221098459
Ridker, P. M., Rifai, N., Rose, L., Buring, J. E., & Cook, N. R. (2002). Comparison of C-Reactive Protein and Low-Density Lipoprotein Cholesterol Levels in the Prediction of First Cardiovascular Events. New England Journal of Medicine, 347(20), 1557–1565. https://doi.org/10.1056/NEJMoa021993
Rott, D., Klempfner, R., Goldenberg, I., & Leibowitz, D. (2015). Cholesterol Levels Decrease soon after Acute Myocardial Infarction . Israel Medical Association Journal, 17(6), 370–373.
Silverman, M. G., Ference, B. A., Im, K., Wiviott, S. D., Giugliano, R. P., Grundy, S. M., Braunwald, E., & Sabatine, M. S. (2016). Association Between Lowering LDL-C and Cardiovascular Risk Reduction Among Different Therapeutic Interventions. JAMA, 316(12), 1289. https://doi.org/10.1001/jama.2016.13985
Siri-Tarino, P. W., Sun, Q., Hu, F. B., & Krauss, R. M. (2010). Meta-analysis of prospective cohort studies evaluating the association of saturated fat with cardiovascular disease. The American Journal of Clinical Nutrition, 91(3), 535–546. https://doi.org/10.3945/ajcn.2009.27725
Sniderman, A. D., Williams, K., Contois, J. H., Monroe, H. M., McQueen, M. J., de Graaf, J., & Furberg, C. D. (2011). A Meta-Analysis of Low-Density Lipoprotein Cholesterol, Non-High-Density Lipoprotein Cholesterol, and Apolipoprotein B as Markers of Cardiovascular Risk. Circulation: Cardiovascular Quality and Outcomes, 4(3), 337–345. https://doi.org/10.1161/CIRCOUTCOMES.110.959247
Tabas, I., Williams, K. J., & Borén, J. (2007). Subendothelial Lipoprotein Retention as the Initiating Process in Atherosclerosis. Circulation, 116(16), 1832–1844. https://doi.org/10.1161/CIRCULATIONAHA.106.676890
Tleyjeh, I. M. (2009). Statins for the Prevention and Treatment of Infections. Archives of Internal Medicine, 169(18), 1658. https://doi.org/10.1001/archinternmed.2009.286
Uthman, O. A., Nduka, C., Watson, S. I., Mills, E. J., Kengne, A. P., Jaffar, S. S., Clarke, A., Moradi, T., Ekström, A.-M., & Lilford, R. (2018). Statin use and all-cause mortality in people living with HIV: a systematic review and meta-analysis. BMC Infectious Diseases, 18(1), 258. https://doi.org/10.1186/s12879-018-3162-1
Wan, Y.-D., Sun, T.-W., Kan, Q.-C., Guan, F.-X., & Zhang, S.-G. (2014). Effect of statin therapy on mortality from infection and sepsis: a meta-analysis of randomized and observational studies. Critical Care, 18(2), R71. https://doi.org/10.1186/cc13828
Wang, B., Guo, Z., Li, H., Zhou, Z., Lu, H., Ying, M., Mai, Z., Yu, Y., Yang, Y., Deng, J., Chen, J., Tan, N., Liu, J., Liu, Y., & Chen, S. (2022). Non-HDL cholesterol paradox and effect of underlying malnutrition in patients with coronary artery disease: A 41,182 cohort study. Clinical Nutrition, 41(3), 723–730. https://doi.org/10.1016/j.clnu.2022.01.027
Wang, N., Woodward, M., Huffman, M. D., & Rodgers, A. (2022). Compounding Benefits of Cholesterol-Lowering Therapy for the Reduction of Major Cardiovascular Events: Systematic Review and Meta-Analysis. Circulation: Cardiovascular Quality and Outcomes, 15(6). https://doi.org/10.1161/CIRCOUTCOMES.121.008552
Wijarnpreecha, K., Panjawatanan, P., Thongprayoon, C., & Ungprasert, P. (2019). Statins & risk of Clostridium difficile infection: A meta-analysis. Indian Journal of Medical Research, 150(4), 359. https://doi.org/10.4103/ijmr.IJMR_1973_17
Williams, K. J., & Tabas, I. (1995). The Response-to-Retention Hypothesis of Early Atherogenesis. Arteriosclerosis, Thrombosis, and Vascular Biology, 15(5), 551–561. https://doi.org/10.1161/01.ATV.15.5.551
Wu, K.-S., Lin, P.-C., Chen, Y.-S., Pan, T.-C., & Tang, P.-L. (2021). The use of statins was associated with reduced COVID-19 mortality: a systematic review and meta-analysis. Annals of Medicine, 53(1), 874–884. https://doi.org/10.1080/07853890.2021.1933165
Yebyo, H. G., Aschmann, H. E., Kaufmann, M., & Puhan, M. A. (2019). Comparative effectiveness and safety of statins as a class and of specific statins for primary prevention of cardiovascular disease: A systematic review, meta-analysis, and network meta-analysis of randomized trials with 94,283 participants. American Heart Journal, 210, 18–28. https://doi.org/10.1016/j.ahj.2018.12.007
Zarny, L. A., & Bernstein, L. H. (1995). Serum Cholesterol: Journal of the American Dietetic Association, 95(9), A25. https://doi.org/10.1016/S0002-8223(95)00436-X
Zein, A. F. M. Z., Sulistiyana, C. S., Khasanah, U., Wibowo, A., Lim, M. A., & Pranata, R. (2022). Statin and mortality in COVID-19: a systematic review and meta-analysis of pooled adjusted effect estimates from propensity-matched cohorts. Postgraduate Medical Journal, 98(1161), 503–508. https://doi.org/10.1136/postgradmedj-2021-140409
Zhang, W., Zhang, Y., Li, C.-W., Jones, P., Wang, C., & Fan, Y. (2017). Effect of Statins on COPD. Chest, 152(6), 1159–1168. https://doi.org/10.1016/j.chest.2017.08.015
kiedy nowe wpisy?
Dobry, Mateuszu.
Lubie Twoje posty, pamiętam że coś skrobałeś na facebooku ale chyba konto usunięte.
Mam kilka zapytań:
1. jak osobiście podchodzisz do kwestii konfliktu interesów w danych badaniach?
jeśli przyjmiemy sceptyczność wobec badań sponsorowanych przez podmioty które mógłby uzyskać korzyść w zależności od wyniku badania, to cała nasza nauka wywraca się o 180 stopni w wielu aspektach
2. czy osobiście przychylasz się do tezy że tłuszcze nasycone są bezwzględnie szkodliwe?
zahaczając o powyższy aspekt, większość meta-analiz zaprzecza temu ale rekomendacje podmiotów zdrowotnych promują taką tezę
tak na początek :)